Introduction
Renal cell carcinoma (RCC) is the most common type
of kidney cancer in adults, responsible for ~80% of cases (1). RCC originates from proximal tubule
epithelial cells. Clear cell renal cell carcinoma (CCRCC) is the
most common subtype of RCC, consisting of 70–80% of metastatic RCC
(2). To date, no sensitive and
specific diagnostic methods have been identified for RCC in
clinical practice. In addition, RCC is relatively resistant to
radiation therapy and chemotherapy (3), which results in a poor prognosis and
low 5-year survival rates among patients with RCC (4). Therefore, future studies on its
pathogenesis are important for the diagnosis and treatment of
RCC.
Previous studies have shown that the von
Hippel-Lindau (VHL) tumor suppressor gene is pivotal in RCC
(5,6). Inactivating mutations of the VHL gene
are a hallmark of CCRCC, since ~60% of CCRCC patients have a
mutated or inactivated VHL gene (7). The wild-type protein encoded by the
VHL gene inhibits the activity of the tumor proliferation proteins
of β-catenin and hypoxia-inducible factor 1 (HIF-1) (5–8). The
β-catenin-mediated Wnt signaling pathway and the HIF-1-activated
Ras-Raf-mitogen-activated protein kinase-extracellular
signal-regulated kinase signaling pathway are associated with cell
proliferation (9). In addition,
HIF-1 upregulates the expression of VEGF and thus, promotes
angiogenesis in the tumor tissue. Therefore, inactivation of the
VHL gene may indirectly relieve the inhibition of the VHL gene on
β-catenin and HIF-1 proteins and thereby promote cell
carcinogenesis. MicroRNAs (miRNAs/miRs) are a class of small
non-coding RNA molecules. Previous studies have demonstrated the
involvement of miRNAs in a wide variety of regulatory pathways,
including development, virus defense, hematopoiesis organogenesis,
cell proliferation, apoptosis and carcinogenesis (10,11).
Compelling evidence has shown that miRNAs may also be involved in
cancer initiation and progression (12,13).
Mature miRNAs are generated via a two-step processing pathway to
yield a 21–25-nucleotide non-coding RNA molecule that regulates
gene expression (14). Mammalian
miRNAs are usually complementary to a site in the 3′ untranslated
region (3′ UTR) (15). miR-30 forms
an miRNA family that includes miR-30a, b, c, d and e, and that has
been identified in humans. Previous observations have shown that
miR-30 is involved in various biological and pathological processes
(16); for example, miR-30a is
required for biliary morphogenesis (17), whereas miR-30c plays a role in
polycythemia vera (18) and cancer
cell drug resistance (19). miR-30d
is localized to chromosomal region 8q24 (20). Previously, a study by Tang et
al (21) reported that miR-30d
may increase glucose-induced insulin gene transcription, but not
the secretion of insulin. Furthermore, the deregulation of miR-30d
in chronic lymphocytic leukemia cells (22) and anaplastic thyroid carcinoma
(23) has also been observed.
Similar to the VHL gene, we hypothesized that miR-30d and its
inactivation or deregulation may also be important in the
pathogenesis of RCC.
Materials and methods
Materials
The renal cancer cells (ACHN cell line) (24) and pcDNA3.1-pre-miR-30d vector were
provided by the Department of Biochemistry and Molecular Biology of
Peking University Health Science Center (Beijing, China).
Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum
were purchased from Gibco (Hangzhou, China), and Lipofectamine 2000
and TRIzol were obtained from Invitrogen Life Technologies
(Carlsbad, CA, USA). Rabbit anti-cyclin E2 was purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The pMIR-REPORT
luciferase vector TaqMan miRNA assay and mirVana qRT-PCR miRNA
detection kits were purchased from Ambion, Inc. (Shanghai, China).
A Roche Light Cycler 480 machine (Roche Diagnostics, Indianapolis,
IN, USA) was used for quantitative polymerase chain reaction
(qPCR). Human RCC specimens were collected following the approval
of histological detection by the Ethics Committee of the Medical
School of Ningbo University (Ningbo, China). All 12 patients
provided written informed consent.
Cell proliferation and colony formation
determination
A colorimetric assay using the tetrazolium salt, MTT
(Sigma-Aldrich, St. Louis, MO, USA), was used to assess cell
proliferation. Equivalent cell numbers (5×103
cells/well) were cultured in 0.2 ml DMEM in each well. Following 1,
2, 3, 4 or 5 days of culture, 20 μl MTT (5 mg/ml) was added to each
well, followed by incubation at 37°C for 3 h. Next, 150 μl dimethyl
sulfoxide was added to solubilize crystals for 10 min. Plates were
immediately read at 540 nm using a microplate reader (KHB ST360;
Shanghai Kehua Bio-engineering Co., Ltd., Shanghai, China). Cell
proliferation curves were obtained using culture days as the
abscissa and MTT absorbance as the ordinate.
For the cell clone formation assay,
~1×103 cells were seeded in a 35-mm cell culture dish
and cultured at 37°C in a 5% CO2 incubator (Thermo
Fisher Scientific, Rockford, IL, USA) for 14 days (until clones
were visible to the naked eye). Following washing with
phosphate-buffered saline (PBS), 1 ml methanol per dish was added
to fix the cells for 15 min. Following staining with crystal violet
dye, cell clone formation was checked under a light microscope
(CKX41; Olympus, Tokyo, Japan).
Cell cycle detection
The cells were seeded in a six-well plate. At 75–80%
confluence, the cells were washed twice with PBS. Following
resuspension in 0.5 ml PBS, the cells were fixed with 4.5 ml
ethanol (70%) overnight. Following centrifugation (200 × g for 10
min), the cells were incubated in a PBS solution containing 0.2
mg/ml propidium iodide, 0.1% Triton X-100 and 0.1 mg/ml RNase at
room temperature, avoiding light for 30 min. Flow cytometry (BD
Biosciences, San Diego, CA, USA) was used to detect the cell cycle.
Cell cycle distribution was analyzed using the ModFit 3.0 program
(Verity Software House, Topsham, ME, USA).
Vector construction
The 3′ UTR region of the cyclin E2 gene was
amplified by qPCR using the total RNA extracted from the ACHN
cells. The following PCR primers were designed: Upstream, 5′-AGA
AGA TAA CTA AGC AAA CAA G-3′; and downstream, 5′-AAT GGG CTA AAA
ATA AAC AGT A-3′. The PCR products were then cloned into the
T-vector. Following confirmation of the target clone sequence by
DNA sequencing, the clone sequence was subcloned into the report
pMIR vector to form a recombinant vector named pMIR3’UTR. For the
mutated vector, the QuikChange II XL site-directed mutagenesis kit
(Stratagene, La Jolla, CA, USA) was used to mutate the seed
sequence on the binding sites for the miR-30d cyclin E2 3′ UTR
(wild-type, 5-TGTTTAC-3; mutant, 5-TGCCCTC-3; underlined sections
indicate the mutated nucleotides). The constructed mutants were
named pMIR3’UTRm1 and pMIR3’UTRm2, respectively.
Plasmid transfection
The cells were seeded and cultured in a 35-mm
culture dish in a 5% CO2 incubator at 37°C until 50%
cell confluence was reached prior to transfection. A DNA mixture
was prepared from varying DNA vectors (10 μg pMIR3’UTR,
pMIR3’UTRm1, pMIR3’UTRm2 or pcDNA-miR30d), plus an equal volume of
Opti-MEM to obtain a final volume of 2.5 ml. The DNA mixtures were
added to centrifuge tubes and incubated at room temperature for 5
min. A liposome solution containing 0.1 ml Lipofectamine 2000 plus
2.4 ml Opti-MEM was prepared and incubated for 5 min. The
DNA-reagent complex was prepared by diluting the DNA mixture with
the liposome solution (1:1 ratio), which was then incubated for 20
min at room temperature. Next, the DNA-reagent complex was added
into the ACHN cell culture medium. Following 8 h of incubation,
culture medium containing the DNA-reagent complex was discarded and
the cells were cultured in normal fresh medium. To obtain stably
transfected cell lines, G418 was used to treat the cells for four
days to select the pcDNA-miR30d clones. For the reporting
measurement, the cells were further cultured for 36 h for the
determination of luciferase activity. Luciferase activity in the
pRL-CMV-transfected cells was used to confirm the transfection
efficiency.
qPCR
Total cellular RNA was extracted following the
TRIzol method and reverse transcribed into cDNA. β-actin was used
as an internal reference. The PCR primers for amplification were as
follows: Cyclin E2 upstream, 5-GCA TTA TGA CAC CAC CGA AGA-3 and
downstream, 5-GGC AAT CAA TCA CAG CAC TAC TT-3; and β-actin
upstream, 5′-AGC GAG CAT CCC CCA AAG TT-3′ and downstream, 5′-GGG
CAC GAA GGC TCA TCA TT-3′. For miR-30d qPCR expression analysis,
the TaqMan miRNA assay and mirVana qRT-PCR miRNA detection kit were
used. U6 small nuclear RNA was used as an internal reference, which
included the following primers: Upstream, 5′-CTCGCTTCGGCAGCACA-3′
and downstream, 5′-AACGCTTCACGAATTTGCGT-3′.
Western blot analysis
Cell lysates (30 μg protein per sample) were diluted
with SDS-PAGE protein sample buffer and then incubated in a 95°C
water bath for 5 min to denature the protein. SDS-polyacrylamide
gel (8%) was used for the electrophoresis. Next, the proteins were
electrically transferred onto a polyvinylidene fluoride (PVDF)
membrane. Subsequent to being washed twice with Tris-buffered
saline with Tween 20 (TBST) buffer, the PVDF membrane was blocked
with 5% skimmed milk in phosphate-buffered saline with Tween 20
(PBST) buffer for 2 h. Next, the membrane was incubated with
primary and secondary antibodies in 5% skimmed milk in PBST buffer
for 2 h successively, washing four times with PBST buffer following
incubation with each antibody. The membrane was developed in
chemiluminescence reagent. Experiments were performed three times
and β-actin expression was used for the control of equal protein
loading.
Statistical analysis
The intergroup differences were tested by Student’s
t-test. P≤0.05 was considered to indicate a statistically
significant difference.
Results
Expression of miR-30d in the RCC
specimens
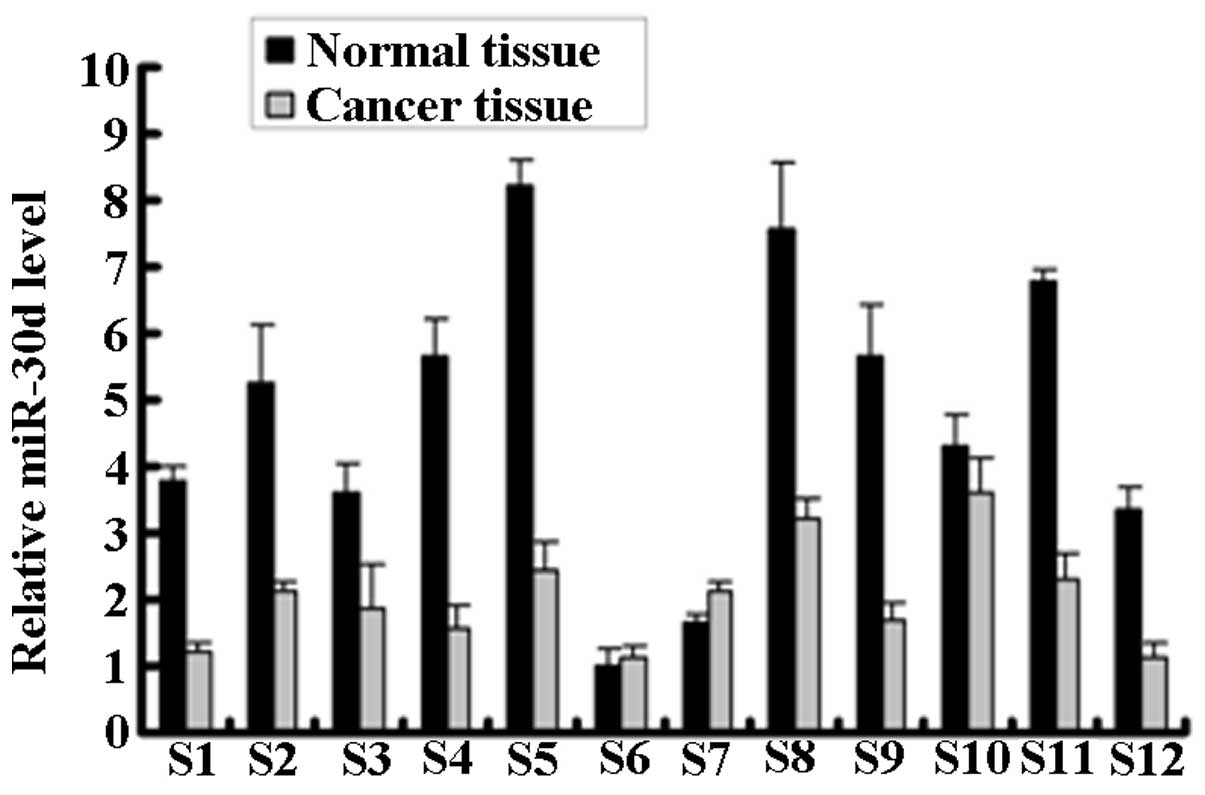
qPCR analysis was used to detect the miR-30d
expression in the renal carcinoma tissue (cancer tissue) and paired
adjacent normal tissue (normal tissue) samples from 12 RCC patients
(Fig. 1). miR-30d expression was
significantly downregulated in the cancer tissues of 9 of the 12
RCC patients.
Proliferation inhibition of miR-30d on
renal carcinoma cells
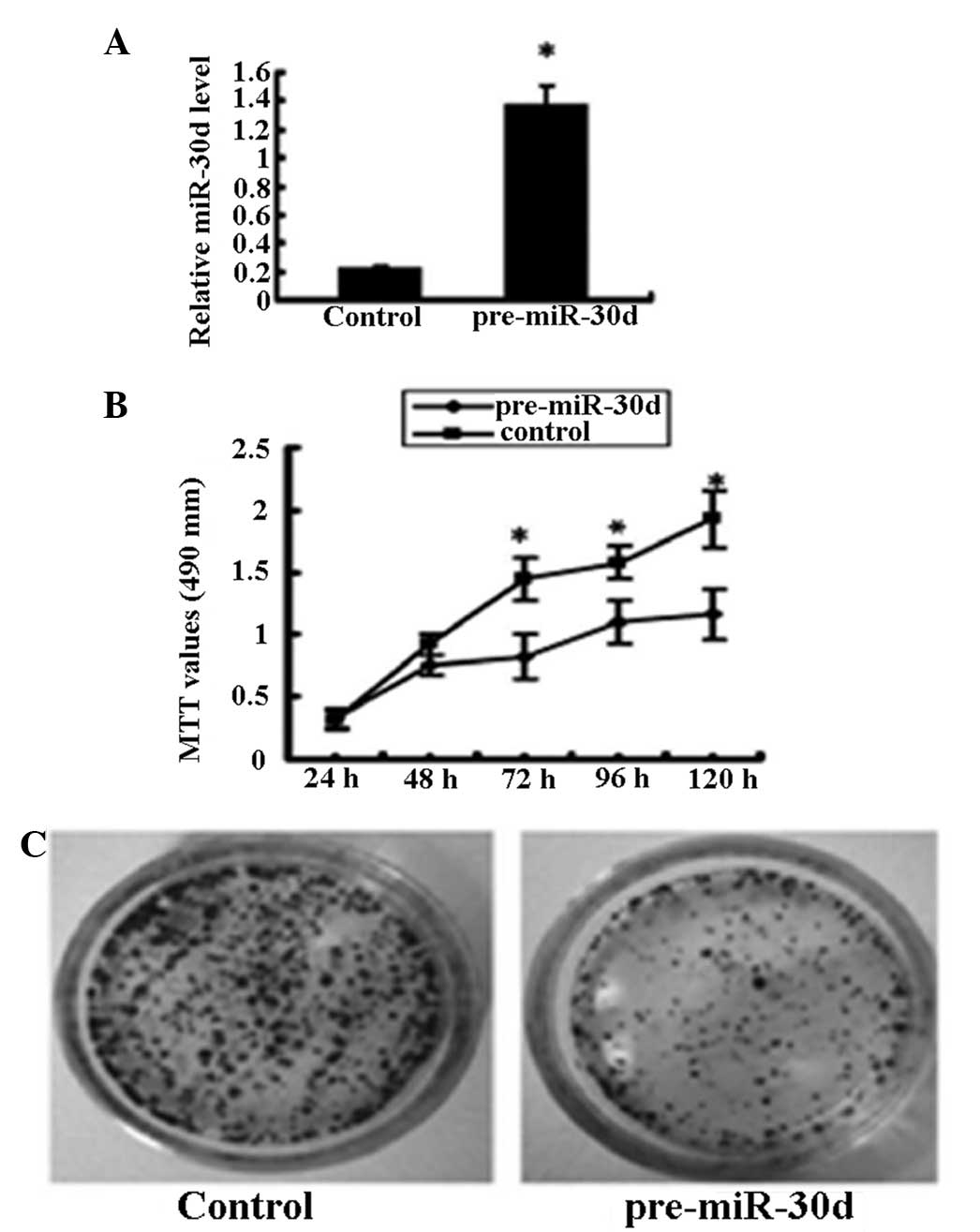
To test the effects of miR-30d on cell
proliferation, renal carcinoma cells (ACHN cell line) that were
untransfected or stably transfected with recombinant plasmid
pcDNA3.1-pre-miR-30d were studied. miR-30d expression in the
pcDNA3.1-pre-miR-30d-transfected cells was upregulated six times as
high as that in the untransfected cells (Fig. 2A). MTT assay showed that
overexpression of miR-30d significantly inhibited renal carcinoma
cell growth (Fig. 2B) and colony
formation (Fig. 2C).
Effects of miR-30d on cyclin E2 at the
post-transcriptional level
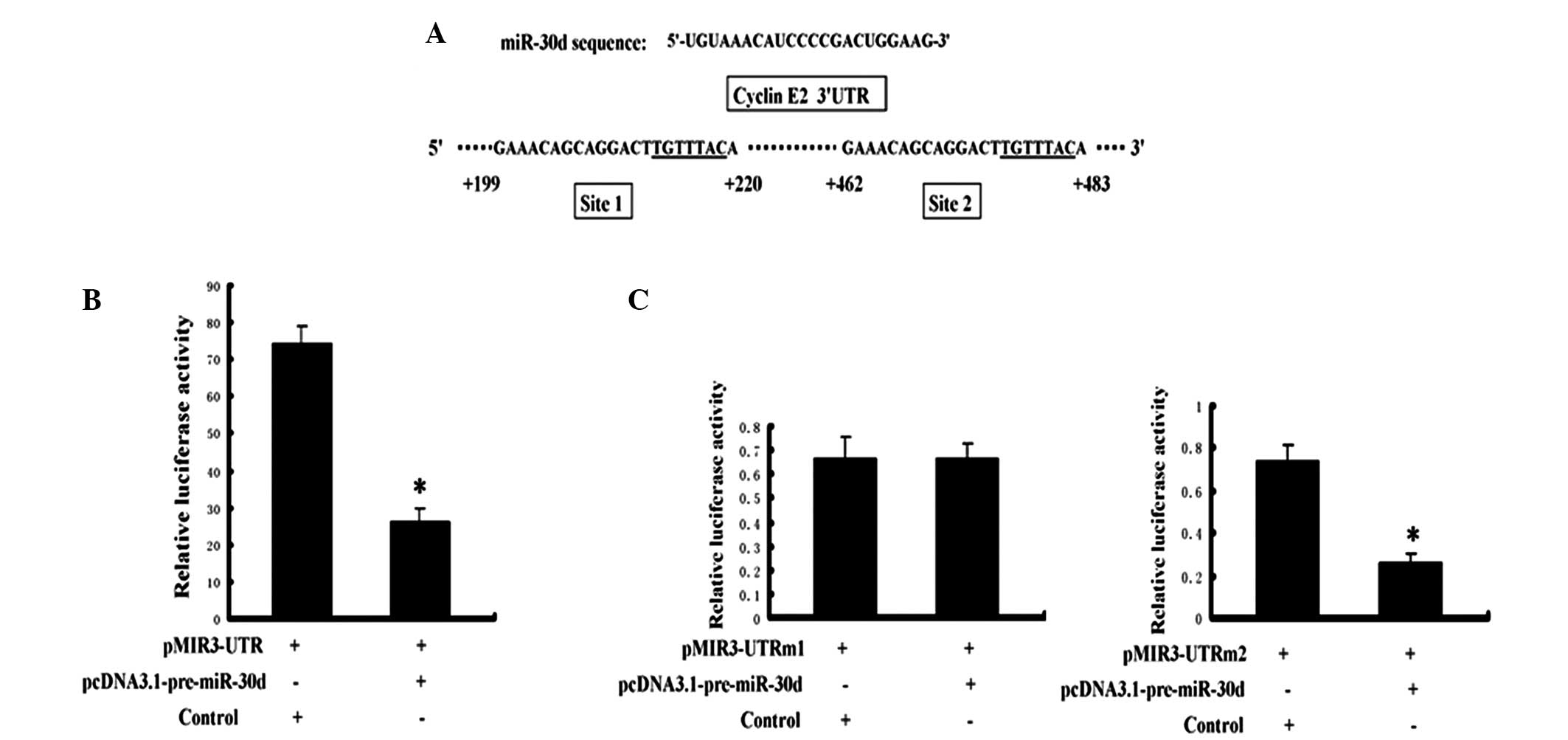
The cyclin E2 gene 3′ UTR reporter vectors,
pMIR3’UTR and pcDNA3.1-pre-miR-30d, were co-transfected into the
renal carcinoma cells. To further confirm that miR-30d directly
regulates cyclin E2, the cyclin E2 3′ UTR and miR-30d were mutated
on the seed sequence of the binding sites. Fig. 3A presents the two miR-30d binding
sites on the cyclin E2 3′ UTR (named site 1 and site 2; underlined
section indicates the seed sequence). The activity of the reporter
gene luciferase was significantly inhibited by miR-30d (Fig. 3B), and the mutation at site 1
significantly attenuated the inhibition of miR-30d (Fig. 3C, left). However, the mutation at
site 2 exhibited no effect on this inhibition (Fig. 3C, right).
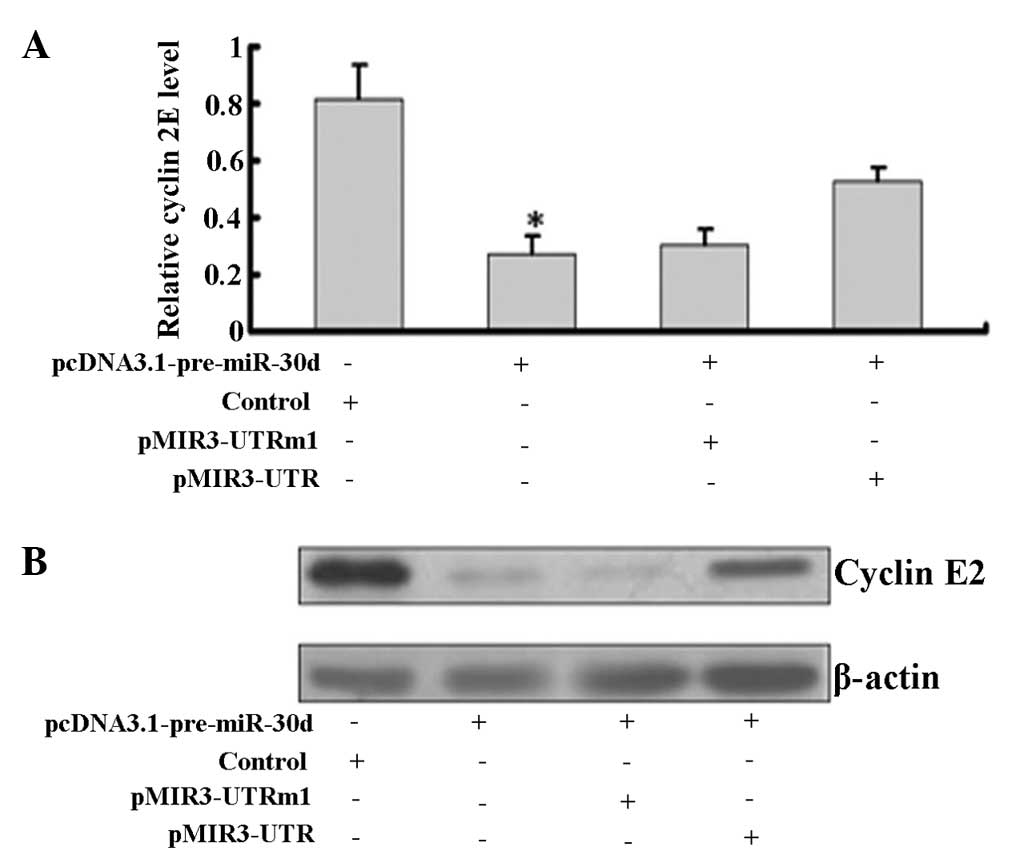
Western blot analysis showed that overexpression of
miR-30d significantly downregulated cyclin E2 at the mRNA and
protein levels in pcDNA3.1-pre-miR-30d-transfected cells (Fig. 4).
Effects of miR-30d on the renal carcinoma
cell cycle
Renal carcinoma cells were collected 48 h after
transfection with pcDNA3.1-pre-miR-30d. The cell cycle was analyzed
using flow cytometry. Compared with the control group, miR-30d
expression significantly increased the content of the cells in the
G1 phase and simultaneously decreased S-phase cell
percentages (Fig. 5).
Discussion
Kidney cancer is one type of urinary system tumor
with a high degree of malignancy, and its pathogenesis remains
unclear. Previous studies have demonstrated that miRNA may be
associated with the occurrence and development of kidney cancer
(25–27). Studies have demonstrated that the
expression of miR-185 in RCC is significantly upregulated (28). miR-185 inhibits the expression of
the tumor suppressor gene, PTEN. PTEN is an important inhibitor of
the phosphoinositide 3-kinase (PI3K)/Akt pathway, and its
downregulation may lead to activation of this signaling pathway
(28,29). An additional study showed that the
miRNA expression of miR-26a, -221/222, -199a-5p, -449a, -21 and
-34a may be associated with RCC (30).
The present study showed that the expression of
miR-30d in renal carcinoma tissue was significantly lower than in
the paired adjacent normal tissue. Conversely, miR-30d
overexpression significantly inhibited the proliferation and colony
formation of renal carcinoma cells in culture, indicating that
miR-30d has the function of a tumor suppressor. Consistent with
this feature, it was further demonstrated that miR-30d binds to
cyclin E2 and inhibits its expression. To confirm this prediction,
the cyclin E2 gene 3′ UTR reporter vectors, pMIR3’UTR and
pcDNA3.1-pre-miR-30d, were co-transfected into the renal carcinoma
cells. The results indicated that the reporter gene luciferase
activity was significantly inhibited by miR-30d, confirming that
cyclin E2 is a target gene of miR-30d. Further experiments
indicated that miR-30d regulates the expression of cyclin E2 by
binding to the 3′ UTR. To test this hypothesis, the cyclin E2 3′
UTR and miR-30d were mutated on the seed sequence of the binding
sites. The results indicated that site 1 mutations attenuate the
inhibition of miR-30d, however, site 2 mutations exhibited no such
inhibitory effect, suggesting that miR-30d regulates the expression
of cyclin E2 by combining with site 1. Further experiments
indicated that the overexpression of miR-30d significantly
downregulates cyclin E2 at the mRNA and protein levels in
pcDNA3.1-pre-miR-30d-transfected cells. In addition, overexpression
of miR-30d may block renal carcinoma cells at the G1
phase, suggesting that cell cycle arrest is an important mechanism
for miR-30d inhibition on the RCC cell proliferation. Cyclin E2 is
a positively regulated factor in the G1 phase of the
cell cycle. The cyclin E2 cyclin-dependent kinase 2, composed of
the serine/threonine kinase complex, regulates the G1
phase of the process. These results indicated that miR-30d inhibits
the expression of cyclin E2, which results in renal carcinoma cell
cycle arrest.
Previously, Esposito et al (31) reported that the downregulation of
miR-30d is associated with the incidence of thyroid cancer. In
addition, Chen et al (32)
found that the overexpression of miR-30d inhibits the proliferation
of HeLa cells. miRNA expression profiling for chronic lymphocytic
leukemia also showed downregulated miR-30d in leukemia cells
(22). These observations are
consistent with the results of the current study, indicating that
miR-30d acts as a tumor suppressor gene. However, certain other
studies have previously indicated that miR-30d may also act in a
similar manner to the oncogenes, since the upregulation of miR-30d
has been reported in hepatocellular carcinoma (16). Kumar et al (33) previously found that miR-30d inhibits
the expression of p53. In addition, the overexpression of miR-30d
genes has been found in medulloblastoma cells (34). These disparities in results may be
due to the difference in cell types, indicating that the exact
roles of miR-30d (acting as an oncogene or tumor suppressor) may be
varied in different cells. Therefore, future studies are necessary
to elucidate the precise molecular mechanism of miR-30d in the
occurrence and development of various types of cancer.
In conclusion, the results of the current study
indicated that miR-30d is important in the regulation of RCC
proliferation. The downregulation of miR-30d may be associated with
the occurrence and development of RCC. Due to the inhibitory
effects of miR-30d on the proliferation of renal carcinoma cells,
this may be a useful new target for the diagnosis and treatment of
kidney cancer.
Acknowledgements
The authors would like to thank Ruth Magaye, Lissy
Lazar and Linda Bowman for their assistance in the preparation of
the present study. The study was partly supported by grants from
the National Nature Science Foundation of China (no. 81273111), the
Foundations of Innovative Research Team of Educational Commission
of Zhejiang Province (no. T200907), the Nature Science Foundation
of Ningbo city (no. 2012A610185), the Ningbo Scientific Project
(nos. 2012C5019 and SZX11073), the Scientific Innovation Team
Project of Ningbo (no. 2011B82014), the Innovative Research Team of
Ningbo (no. 2009B21002) and the Wong Magna Fund in Ningbo
University.
References
|
1
|
Farber LJ, Furge K and Teh BT: Renal cell
carcinoma deep sequencing: recent developments. Curr Oncol Rep.
14:240–248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reuter VE: The pathology of renal
epithelial neoplasms. Semin Oncol. 33:534–543. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Itsumi M and Tatsugami K: Immunotherapy
for renal cell carcinoma. Clin Dev Immunol. 2010:2845812010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walsh N, Larkin A, Kennedy S, et al:
Expression of multidrug resistance markers ABCB1 (MDR-1/P-gp) and
ABCC1 (MRP-1) in renal cell carcinoma. BMC Urol. 9:62009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Linehan WM, Rubin JS and Bottaro DP: VHL
loss of function and its impact on oncogenic signaling networks in
clear cell renal cell carcinoma. Int J Biochem Cell Biol.
41:753–756. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Linehan WM, Pinto PA, Srinivasan R, et al:
Identification of the genes for kidney cancer: opportunity for
disease-specific targeted therapeutics. Clin Cancer Res.
13:671s–679s. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
An J and Rettig MB: Mechanism of von
Hippel-Lindau protein-mediated suppression of nuclear factor kappa
B activity. Mol Cell Biol. 25:7546–7556. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Peruzzi B, Athauda G and Bottaro DP: The
von Hippel-Lindau tumor suppressor gene product represses oncogenic
β-catenin signaling in renal carcinoma cells. Proc Natl Acad Sci
USA. 103:14531–14536. 2006.PubMed/NCBI
|
|
9
|
Conrad PW, Freeman TL, Beitner-Johnson D
and Millhorn DE: EPAS1 trans-activation during hypoxia requires
p42/p44 MAPK. J Biol Chem. 274:33709–33713. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carrington JC and Ambros V: Role of
microRNAs in plant and animal development. Science. 301:3362003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yanaihara N, Caplen N, Bowman E, et al:
Unique microRNA molecular profiles in lung cancer diagnosis and
prognosis. Cancer Cell. 9:189–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Horikawa Y, Wood CG, Yang H, et al: Single
nucleotide polymorphisms of microRNA machinery genes modify the
risk of renal cell carcinoma. Clin Cancer Res. 14:7956–7962. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Trang P, Weidhaas J and Slack F: MicroRNAs
as potential cancer therapeutics. Oncogene. 27(Suppl 2): S52–S57.
2008. View Article : Google Scholar
|
|
15
|
Didiano D and Hobert O: Perfect seed
pairing is not a generally reliable predictor for miRNA-target
interactions. Nat Struct Mol Biol. 13:849–851. 2006. View Article : Google Scholar
|
|
16
|
Yao J, Liang L, Huang S, et al:
MicroRNA-30d promotes tumor invasion and metastasis by targeting
Galphai2 in hepatocellular carcinoma. Hepatology. 51:846–856.
2010.PubMed/NCBI
|
|
17
|
Hand NJ, Master ZR, Eauclaire SF,
Weinblatt DE, Matthews RP and Friedman JR: The microRNA-30 family
is required for vertebrate hepatobiliary development.
Gastroenterology. 136:1081–1090. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bruchova H, Merkerova M and Prchal JT:
Aberrant expression of microRNA in polycythemia vera.
Haematologica. 93:1009–1016. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sorrentino A, Liu CG, Addario A, Peschle
C, Scambia G and Ferlini C: Role of microRNAs in drug-resistant
ovarian cancer cells. Gynecol Oncol. 111:478–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schlaeger C, Longerich T, Schiller C, et
al: Etiology-dependent molecular mechanisms in human
hepatocarcinogenesis. Hepatology. 47:511–520. 2008. View Article : Google Scholar
|
|
21
|
Tang X, Muniappan L, Tang G and Ozcan S:
Identification of glucose-regulated miRNAs from pancreatic (beta)
cells reveals a role for miR-30d in insulin transcription. RNA.
15:287–293. 2009. View Article : Google Scholar
|
|
22
|
Marton S, Garcia MR, Robello C, et al:
Small RNAs analysis in CLL reveals a deregulation of miRNA
expression and novel miRNA candidates of putative relevance in CLL
pathogenesis. Leukemia. 22:330–338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Visone R, Pallante P, Vecchione A, et al:
Specific microRNAs are downregulated in human thyroid anaplastic
carcinomas. Oncogene. 26:7590–7595. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu C, Jin B, Chen L, et al: MiR-30d
induces apoptosis and is regulated by the Akt/FOXO pathway in renal
cell carcinoma. Cell Signal. 25:1212–1221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Petillo D, Kort EJ, Anema J, Furge KA,
Yang XJ and Teh BT: MicroRNA profiling of human kidney cancer
subtypes. Int J Oncol. 35:109–114. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nakada C, Matsuura K, Tsukamoto Y, et al:
Genome-wide microRNA expression profiling in renal cell carcinoma:
significant down-regulation of miR-141 and miR-200c. J Pathol.
216:418–427. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Juan D, Alexe G, Antes T, et al:
Identification of a microRNA panel for clear-cell kidney cancer.
Urology. 75:835–841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pantuck AJ, Seligson DB, Klatte T, et al:
Prognostic relevance of the mTOR pathway in renal cell carcinoma.
Cancer. 109:2257–2267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rathmell WK and Chen S: VHL inactivation
in renal cell carcinoma: implications for diagnosis, prognosis and
treatment. Expert Rev Anticancer Ther. 8:63–73. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Redova M, Svoboda M and Slaby O: MicroRNAs
and their target gene networks in renal cell carcinoma. Biochem
Biophys Res Commun. 405:153–156. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Esposito F, Tornincasa M, Pallante P, et
al: Down-regulation of the miR-25 and miR-30d contributes to the
development of anaplastic thyroid carcinoma targeting the polycomb
protein EZH2. J Clin Endocrinol Metab. 97:E710–E718. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen LZ, Niu XH, Han XL, Xie BS and Mao
ZB: Down-regulation of miR-30d on proliferation of HeLa cells.
Chinese Journal of Biochemistry and Molecular Biology.
11:1047–1052. 2009.
|
|
33
|
Kumar M, Lu Z, Takwi AA, et al: Negative
regulation of the tumor suppressor p53 gene by microRNAs. Oncogene.
30:843–853. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu Y, Ryan SL, Elliott DJ, et al:
Amplification and overexpression of Hsa-miR-30b, Hsa-miR-30d and
KHDRBS3 at 8q24.22–q24.23 in medulloblastoma. PLoS One.
4:e61592009.PubMed/NCBI
|