Introduction
Desmoplastic small round cell tumor (DSRCT), which
was first described by Gerald and Rosai (1), is a rare, high grade and aggressive
malignant tumor, defined as a mesenchymal neoplasm that grows along
serosal surfaces. DSRCT often affects the abdominal and/or pelvic
peritoneum, predominantly affecting young adult males (2). Despite aggressive combination
interventions, such as poly-chemotherapy, debulking surgery and
whole abdominal radiation, the therapeutic management of DSRCT
remains unsatisfactory and subsequently has a poor patient
prognosis (3). Surgical resection
is only recommended for non-metastatic disease with combination
chemoradiotherapy as an adjunct, however, the outcome is often
unefficacious, predominantly due to disease recurrence. For
patients in the advanced stages of the disease, symptom control is
crucial as the aforementioned interventions only marginally impact
survival, thus, palliation of secondary symptoms is of paramount
importance (4). Furthermore, it is
essential to decide on appropriate subsequent treatments as
currently, there is no consensus on any such strategy. Therefore,
in the present study, a patient with recurrent and metastatic DSRCT
is described, who received surgical resection in combination with
chemotherapy, but was successfully treated by the surgery alone.
Patient provided written informed consent.
Case report
In May 2010, 39-year-old Iranian male visited the
Ibaraki Medical Center, Tokyo Medical University Hospital (Ami,
Japan) presenting with lower abdominal and pelvic pain together
with constipation. His medical records revealed that he had
undergone palliative surgery at another hospital for a large
intra-abdominal tumor two years previously. Histological diagnosis
of the resected specimen indicated DSRCT. One year after the first
surgery, the residual tumor had grown rapidly and he had received
anticancer medications. However, the residual tumor appeared to be
worsening and the patient complained of debilitating symptoms.
Physical examinations on the individuals’ admission to the Ibaraki
Medical Center, Tokyo Medical University Hospital revealed lower
abdominal tenderness without a palpable mass. The majority of the
laboratory assessment results were normal, with the exception of a
slight liver function disorder. Furthermore, numerous tumorigenic
factors, including carcinoembryonic antigen, carbohydrate antigen
(CA) 125, CA 19-9 and neuron specific enolase (NSE) were within the
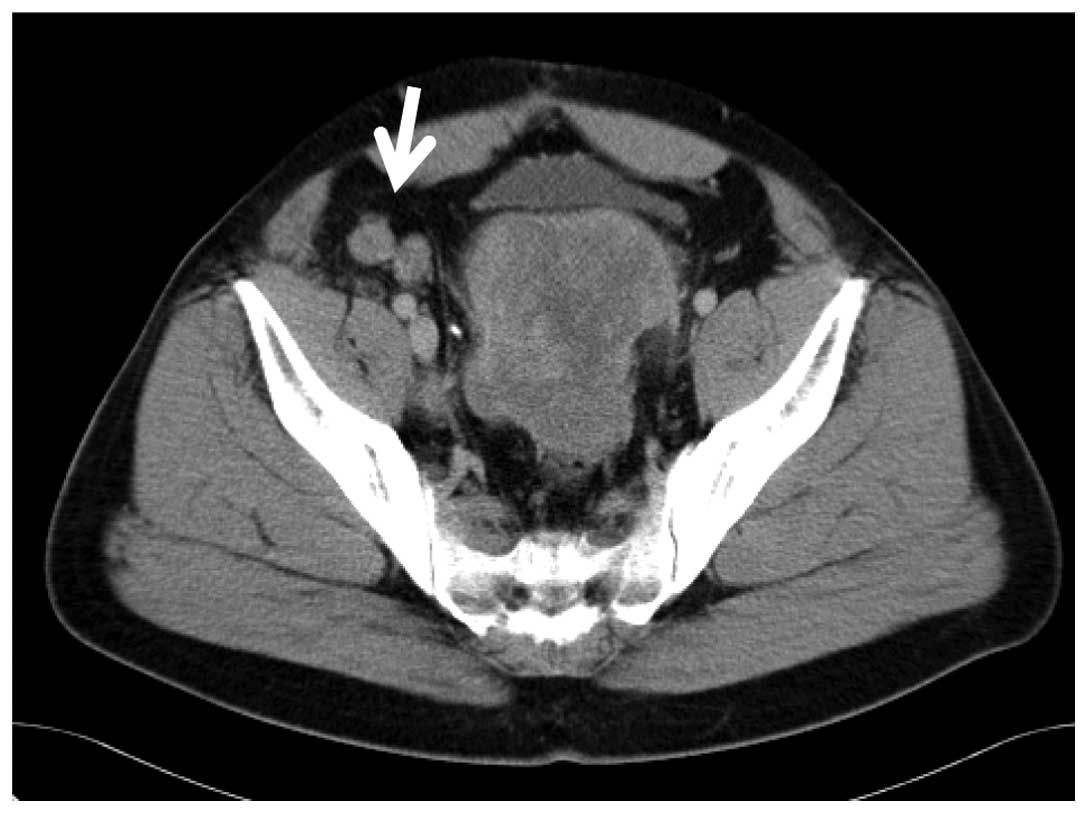
normal range. Abdominal enhanced computed tomography (CT) revealed
a large mass (size, 18×7 cm) with slight heterogeneous enhanced
areas in the pouch of Douglas, coupled with nodules in the
abdominal cavity (Fig. 1). In
magnetic resonance imaging (MRI), the large mass in the pouch of
Douglas appeared hypointense in the T1-weighted images and
heterogeneous hyperintense in the T2-weighted images (Fig. 2). In addition, the mass had
compressed the rectosigmoid colon and the bladder. The diagnosis
was, therefore, metastasis and recurrence of a DSRCT that was
growing rapidly.
Among the several treatment strategies available,
the patient opted for surgical excision of the tumor. A laparotomy
was performed to relieve the patient’s symptoms, during which a
voluminous mass was identified that had occupied the pouch of
Douglas. Furthermore, numerous small nodules were observed on the
omentum, which were coupled with diffuse peritoneal seeding on the
surface of the diaphragm. The large mass had penetrated the
anterior wall of the rectum, however, the bladder and the urethra
were not affected. The tumor was removed via a low anterior
resection and a covering ileostomy was constructed. However,
complete elimination of the tumor appeared to be difficult due to
multiple peritoneal metastases, spreading to the nodules, diaphragm
and spleen.
Macroscopically, the tumor, which measured 18×11×7
cm, had an uneven surface and when cut, necrotic areas were
observed. Histological examinations of the excised tumor revealed
that the tumor cells had focally invaded the muscularis propria of
the rectum (Fig. 3A). Invasive
small round or short spindle cells were identified embedded in the
desmoplastic stroma (Fig. 3B). The
tumor cells had round to oval nuclei with increased chromatins,
which were accompanied by eosinophilic cytoplasm with numerous
mitotic features. Foci of necrosis and vascular permeation were
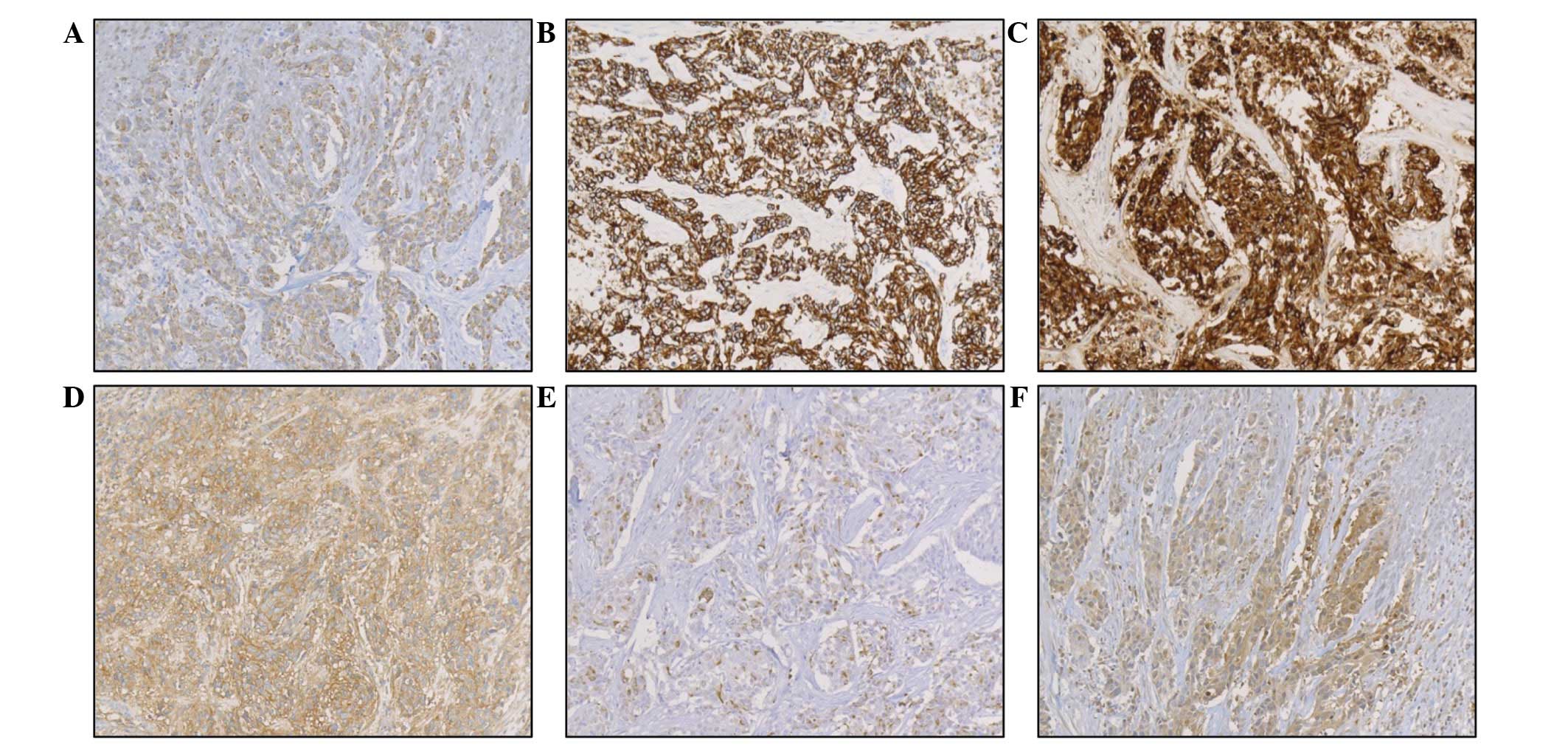
frequently observed. Immunohistochemical investigations revealed
positive staining for cytokeratin (CK) AE1/AE3, CK CAM5.2,
epithelial membrane antigen (EMA), cluster of differentiation
(CD)99, desmin and NSE (Fig. 4),
but negative staining for CK 34βE12, CK 5/6, CK 7, CK 20, c-kit,
CD34, S-100 protein, CA 125, neurofilament, synaptophysin and
α-smooth muscle actin (α-SMA). The MIB-1 index was 70% and focal
positive staining for Wilms tumor-1 (WT1) was also noted. These
immunohistological results supported the diagnosis of a progressing
DSRCT. The postoperative period was uneventful and the patient was
discharged 14 days later.
The patient was followed up and one year later,
complained of intermittent pain in the right lower limbs. A
follow-up examination by CT scan revealed metastasis and growth of
tumor cells in the inguinal lymph nodes. A right inguinal
lymphadenectomy was performed and five days later, the patient was
discharged with no postoperative complications. The excised lymph
nodes measured 4.5×3.0 and 5.0×2.0 cm. In the immunohistochemical
examinations of the excised lymph nodes, CK AE1/AE3, CK CAM5.2,
EMA, CD99, desmin, NSE, CK 7, c-kit and CA 125 were positive,
however, CK 34βE12, CK 5/6, CK 20, CD34, S-100 protein,
neurofilament, synaptophysin and α-SMA were negative. The MIB-1
index was 70% and these pathological findings of the lymph nodes
were compatible with the metastatic DSRCT. At the present time, 60
months following the first diagnosis of DSRCT, the patient
continues to be symptom free, without any local progression of the
tumor.
Discussion
DSRCT is a rare, but aggressive type of tumor with a
poor prognosis and a high prevalence in young males, with the peak
age at diagnosis ranging between 16 and 26 years (5,6). In
the majority of cases, DSRCT appears as a large mass in the
abdominal cavity with serosal and omental spreading, and rapidly
metastasizes to the liver, lungs, lymph nodes and the peritoneum
(7,8). The most common symptoms of DSRCT are
non-specific. The majority of patients present with a palpable mass
in the abdominal cavity, coupled with pelvic lesions, and the
associated symptoms include abdominal pain, constipation, weight
loss and distension (7).
Accordingly, the patient in the present case complained of lower
abdominal and pelvic pain with impaired bowel movement. Imaging
techniques, including MRI and CT scans, are indispensible as tools
for diagnosis, identification of tumor location and assessment of
tumor progression. An intra-abdominal DSRCT often appears as
multiple bulky, lobulated and heterogeneous masses, with hypodense
areas in unenhanced CT images, and weak heterogeneous areas in
contrast-enhanced CT images (5,9).
Malignant peritoneal mesothelioma, rhabdomyosarcoma and lymphoma
may demonstrate a radiographic appearance similar to DSRCT and
should therefore be considered for differential diagnosis (10). In the present case, abdominal
enhanced CT scan revealed that the large mass with weak
heterogeneous enhanced areas in the pouch of Douglas, involved the
rectosigmoid colon and a number of nodules (or seeding) in the
abdominal cavity, in addition to metastasis to the inguinal lymph
nodes. No calcification or heterogeneous hypodense areas were
identified. In MRI examinations, DSRCT often presents as lesions
with heterogeneous iso- or hypointense areas in T1-weighted MR
images and heterogeneous hyperintense in T2-weighted MR images
(10,11). In the present case, the large mass
of the pouch of Douglas appeared hypointense in T1-weighted images
and heterogeneous hyperintense in T2-weighted images.
DSRCT is a member of the family of malignant small
round cell tumors. They are characterized by small, round,
relatively undifferentiated cells and generally include Ewing’s
sarcoma, peripheral neuroectodermal tumor, rhabdomyosarcoma,
synovial sarcoma, non-Hodgkin’s lymphoma, retinoblastoma,
neuroblastoma, hepatoblastoma, and nephroblastoma or Wilms tumor.
Differential diagnosis of small round cell tumors is particularly
difficult due to their undifferentiated or primitive features
(12). Histopathologically, the
majority of tumors exhibit a nesting or solid/diffuse pattern, with
a characteristic histological appearance of high cellularity,
undifferentiated small to medium size uniform round cells and a
sparse cytoplasm. Nuclei are round to oval shaped and
morphologically arranged in nest or spindle cell formation,
embedded in dense and metachromatic desmoplastic stroma (13–15).
The cells have high nuclear/cytoplasmic ratios with granular
chromatin that is reminiscent of small cell carcinoma and
pseudorosettes are observed in certain specimens. These features
are key to the diagnosis of DSRCT. Currently, the diagnosis of
DSRCT is based on immunohistochemical and molecular analysis, which
are used as tools for the confirmation of diagnosis. Furthermore,
DSRCT is a unique tumor with multiple phenotypic differentiations
and characteristic immunohistochemical features. These features
reflect diversity in the morphology of DSRCT, exhibiting
characteristic features with regard to the epithelium, muscles and
nerves. Previous studies have demonstrated that DSRCT exhibited
strong and diffuse cytoplasmic immunoreactivity for CK AE1/AE3,
vimentin, desmin, NSE and EMA, however, S-100 protein, chromogranin
A, cynaptophysin and neurofilament were negative or weak (16–18).
In the present case, positive immunoreactivity was observed for CK
AE1/AE3, CK CAM5.2, EMA, CD99, desmin, NSE and WT1, but were
negative for CK 34βE12, CK 5/6, CK 7, CK 20, c-kit, CD34, S-100
protein, CA 125, neurofilament, synaptophysin and α-SMA. The latter
results were not consistent with a diagnosis of DSRCT.
The prognosis of patients with DSRCT remains poor
and despite the availability of therapeutic strategies, including
surgical resection combined with radiotherapy and chemotherapy, the
mortality rates remain high. Although surgery, chemotherapy,
radiotherapy and combined therapy have been used in the treatment
of DSRCT, no single therapy has been accepted as the standard
strategy. Complete excision is often difficult due to the presence
of multiple or diffuse metastases in the peritoneum. In general,
the patients with metastasis have a poor prognosis even following
chemotherapy and/or radiotherapy. In a review of 66 patients with
DSRCT, Lal et al (7)
reported that the overall three and five-year survival rates were
44 and 15%, respectively. Gross tumor resection was highly
significant in overall survival as the three-year survival rate in
patients that were treated by gross tumor resection was 58%.
Debulking surgery is attempted with a goal of ≥90% resection of the
tumor bulk and aggressive surgical resection continues to be a
major determinant of patient survival. Gil et al (17) reported that the median survival time
of patients with complete cytoreduction was 20 months (range, 13–55
months). In another study, the 12 patients with DSRCT were reviewed
and the median survival time of patients who underwent surgical
resection compared with those who underwent biopsy alone was 34
versus 14 months, respectively (6).
In 7/12 patients, surgical resection was attempted, however,
macroscopic total resection of the tumor was accomplished in 3/7
patients and the remaining four patients underwent major debulking
surgery. All patients who underwent macroscopic total resection
subsequently developed recurrence, which required additional
surgery.
Tumor recurrence and progression is common in
patients with DSRCT. Accordingly, decisions regarding the
appropriate follow-up treatment strategy are essential. Aggressive
surgery combined with multi-agent adjuvant chemotherapy is
recommended to relieve symptoms and to improve the outcome.
Numerous aggressive combination chemotherapy protocols, including
IRS-38 (oncovin, platinol, adriamycin, cyclophosphamide), VAC
(oncovin, endoxan, actinomycin-D), IVA (ifosfamide, vincristine,
adriamycin), P6 (cyclophosphamide, doxorubicin, vincristine,
ifosfamide, etoposide) and PAVEP (cyclophosphamide, etoposide,
doxorubicin, cisplatin), have been attempted with a certain degree
of chemosensitivity and improved survival rates (19–22).
However, during aggressive chemotherapy, drug toxicity may be
severe and often requires hospitalization. Regarding a standard
strategy for the treatment of DSRCT, neoadjuvant chemotherapy,
>90% tumor debulking and radiotherapy have been demonstrated to
prolong survival (4). Aggressive
surgical resection of extensive intra-abdominal DSRCT correlates
with an improvement of therapeutic outcomes (5). In the present case, the patient
underwent surgical resection of a large abdominal tumor prior to
admission to our hospital, however, the tumors had evidently not
been removed completely because numerous nodules in the abdominal
cavity were observed. Although chemotherapy had been applied in
another hospital, the patient’s tumor was progressing upon our
first examination. The patient refused additional chemotherapy and
was followed up. One year later, the residual tumor had grown
rapidly regardless of the surgery. To reduce the patient’s
symptoms, debulking surgery was performed twice. At the time of
writing, the patient continues to survive with no symptoms and the
initial surgery was 60 months ago. Therefore, it was hypothesized
that surgical debulking relieves symptoms and improves survival
time in metastatic and recurrent DSRCT patients. Recent efforts
have focused on improving disease control without increasing
treatment-associated morbidity (23).
In conclusion, this case report presents a potential
treatment strategy for patients who develop recurrence of
intra-abdominal DSRCT as a solitary mass with multiple seeding of
the peritoneum.
References
|
1
|
Gerald WL and Rosai J: Case 2.
Desmoplastic small round cell tumor with divergent differentiation.
Pediatr Pathol. 9:177–183. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gerald WL, Miller HK, Battifora H,
Miettinen M, Silva EG and Rosai J: Intra-abdominal desmoplastic
small round-cell tumor. Report of 19 cases of a distinctive type of
high-grade polyphenotypic malignancy affecting young individuals.
Am J Surg Pathol. 15:499–513. 1991. View Article : Google Scholar
|
|
3
|
Dufresne A, Cassier P, Couraud L,
Marec-Berard P, Meeus P, Alberti L and Blay JY: Desmoplastic small
round cell tumor: current management and recent findings. Sarcoma.
2012:7149862012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stuart-Buttle CE, Smart CJ, Pritchard S,
Martin D and Welch IM: Desmoplastic small round cell tumour: a
review of literature and treatment options. Surg Oncol. 17:107–112.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chouli M, Viala J, Dromain C, Fizazi K,
Duvillard P and Vanel D: Intra-abdominal desmoplastic small round
cell tumors: CT findings and clinicopathological correlations in 13
cases. Eur J Radiol. 54:438–442. 2005. View Article : Google Scholar
|
|
6
|
Hassan I, Shyyan R, Donohue JH, Edmonson
JH, Gunderson LL, Moir CR, Arndt CA, Nascimento AG and Que FG:
Intraabdominal desmoplastic small round cell tumors: a diagnostic
and therapeutic challenge. Cancer. 104:1264–1270. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lal DR, Su WT, Wolden SL, Loh KC, Modak S
and La Quaglia MP: Result of multimodal treatment for desmoplastic
small round cell tumors. J Pediatr Surg. 40:251–255. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mrabti H, Kaikani W, Ahbeddou N, Abahssain
H, El Khannoussi B, Amrani M and Errihani H: Metastatic
desmoplastic small round cell tumor controlled by an
anthracycline-based regimen: review of the role of chemotherapy. J
Gastrointest Cancer. 43:103–109. 2012. View Article : Google Scholar
|
|
9
|
Zhang WD, Li CX, Liu QY, Hu YY, Cao Y and
Huang JH: CT, MRI, and FDG-PET/CT imaging findings of
abdominopelvic desmoplastic small round cell tumors: correlation
with histopathologic findings. Eur J Radiol. 80:269–273. 2011.
View Article : Google Scholar
|
|
10
|
Kis B, O’Regan KN, Agoston A, Javery O,
Jagannathan J and Ramaiya NH: Imaging of desmoplastic small round
cell tumour in adults. Br J Radiol. 85:187–192. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tateishi U, Hasegawa T, Kusumoto M, Oyama
T, Ishikawa H and Moriyama N: Desmoplastic small round tumor:
imaging findings associated with clinicopathologic featres. J
Comput Assist Tomogr. 26:579–583. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rajwanshi A, Srinivas R and Upasana G:
Malignant small round cell tumors. J Cytol. 26:1–10. 2009.
View Article : Google Scholar
|
|
13
|
Baz W, EI-Soueidi R, Nakhl F, Aoun N, Chin
N and Dhar M: Desmoplastic small round-cell tumor: an adult with
previous exposure to agent orange. Jpn J Clin Oncol. 40:593–595.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao L, Ni J, Que R, Wu Z and Song Z:
Desmoplastic small round cell tumor: a clinical, pathological, and
immunohistochemical study of 18 Chinese cases. Int J Surg Pathol.
16:257–262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Crapanzano JP, Cardillo M, Lin O and
Zakowski MF: Cytology of desmoplastic small round cell tumor.
Cancer. 96:21–31. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dorsey BV, Benjamin LE, Rauscher F 3rd,
Klencke B, Venook AP, Warren RS and Weidner N: Intra-abdominal
desmoplastic small round-cell tumor: expansin of the pathologic
profile. Mod Pathol. 9:703–709. 1996.PubMed/NCBI
|
|
17
|
Gil A, Gomez Portilla A, Brun EA and
Sugarbaker PH: Clinical perspective on desmoplastic small
round-cell tumor. Oncology. 67:231–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rekhi B, Ahmed S, Basak R, Qureshi SS,
Desai SS, Ramadwar M, Desai SB, kurkure P and Jambhekar NA:
Desmoplastic small round cell tumor-clinicopathological spectrum,
including unusual features and immunohistochemical analysis of 45
tumors diagnosed at a tertiary cancer referral centre, with
molecular results t(11; 22) (p13; q12) (EWS-WT1) in select cases.
Pathol Oncol Res. 18:917–927. 2012. View Article : Google Scholar
|
|
19
|
Livaditi E, Mavridis G, Soutis M,
Papandreou E, Moschovi M, Papadakis V, Stefanaki K and
Christopoulos-Geroulanos G: Diffuse intraabdominal desmoplastic
small round cell tumor: a ten-year experience. Eur J Pediatr Surg.
16:423–427. 2006.PubMed/NCBI
|
|
20
|
Kurre P, Felgenhauer JL, Miser JS,
Patterson K and Hawkins DS: Successful dose-intensive treatment of
desmoplastic small round cell tumor in three children. J Pediatr
Hematol Oncol. 5:446–50. 2000. View Article : Google Scholar
|
|
21
|
Farhat F, Culine S, Lhommé C, Duvillard P,
Soulié P, Michel G, Terrier-Lacombe MJ, Théodore C, Schreinerova M
and Droz JP: Desmoplastic small round cell tumors: results of a
four-drug chemotherapy regimen in five adult patients. Cancer.
77:1363–1366. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Biswas G, Laskar S, Banavali SD, Gujral S,
Kurkure PA, Muckaden M, Parikh PM and Nair CN: Desmoplastic small
round cell tumor: extra abdomimal and abdominal presentations and
the results of treatment. Indian J Cancer. 42:78–84. 2005.
View Article : Google Scholar
|
|
23
|
Pinnix CC, Fontanilla HP, Hayes-Jordan V,
Subbiah V, Bilton SD, Chang EL, Grosshans DR, McAleer MF, Sulman
EP, Woo SY, Anderson P, Green HL and Mahajan A: Whole
abdominopelvic intensity-modulated radiation therapy for
desmoplastic small round cell tumor after surgery. Int J Radiat
Oncol Biol Phys. 83:317–326. 2012. View Article : Google Scholar
|