Introduction
Langerhans cell histiocytosis (LCH) is a rare
disease of unknown etiology characterized by mixed cellular
infiltration with colonial proliferation of LCs in specific
histopathological lesions, which results in a variety of clinical
manifestations. LCs are identical to normal dendritic cells. These
atypical and immature cells of the mononuclear phagocytic system
can infiltrate virtually anywhere in the body and may occur in
localized lesions or as widespread systemic diseases (1). LCH has an extremely variable
presentation that depends on abnormal proliferation and
dissemination of histiocytes. Therefore, LCH has numerous clinical
forms that affect different systems or different sites in the same
system with variable outcomes (2).
LCH occurs in the bones, skin and mucous membranes in children, and
occasionally in other organs in adults.
Depending on the extent and localization of the
disease at the time of evaluation, LCH can be classified as single
system LCH when one organ/system is involved or multisystem LCH
(MS-LCH) when two or more organs/systems are involved. MS-LCH is
reported in <30% of LCH cases (3). The hematopoietic system, liver and/or
spleen are considered high-risk organs rich in histiocytes. LCH has
been reported in two to five cases per million individuals annually
and rarely occurs in adults with liver or pituitary lesions
(1). This study reports an adult
LCH patient with liver and pituitary dysfunction as well as spleen
damage. To the best of our knowledge, manifestation of LCH in the
liver, spleen and pituitary gland of an adult has not been reported
previously.
Diagnosis of LCH is based on histological and
immunophenotypic examination of lesional tissue. The key step is
the morphological identification of the characteristic LCs.
Positive cluster of differentiation (CD)1a and/or Langerin (CD207)
staining in lesional cells is required for definitive diagnosis
(4). LCH has variable clinical
symptoms, as it affects a wide variety of systems (2). Specific symptoms include pain,
swelling, skin rashes, otorrhea, irritability, fever, loss of
appetite, diarrhea, polydipsia, dyspnea and behavioral and
neurological changes (5).
Currently, there is no standard treatment for LCH. Age, extent of
disease and dysfunction of vital organs are major factors that need
to be considered in deciding treatment. Due to the fact that no
previous standard of care exists for the treatment of LCH, the
Histiocyte Society developed the Histiocyte Society Evaluation and
Treatment Guidelines in April, 2009 (6). According to the guidelines, LCH
patients with multiorgan involvement and dysfunction of the liver,
lungs or bone marrow are considered a high-risk group. An initial
six-week course of therapy with vinblastine and prednisone is
recommended for all patients with multisystem disease. Further
therapy depends on patient response to the initial therapy. Written
informed consent was obtained from the patient.
Case report
A 45-year-old male visited the Second Xiangya
Hospital (Changsha, China) with complaints of fever, fatigue,
anorexia, jaundice and polyuria for two months. Physical
examination revealed that body temperature and heart rate were
38.3°C and 95 bpm, respectively, with moderately stained skin and
sclera. The spleen was found to be enlarged 3 cm below the costal
margin. The patient exhibited slight pain in the region of the
liver. The patient had no bone pain or superficial lymphadenopathy.
No abnormalities were detected on heart and lung examination.
Laboratory testing revealed significantly elevated
levels of alanine transaminase (200 U/l; normal range, 0–40 U/l),
aspartate transaminase (256 U/l; normal range, 0–37 U/l), total
bilirubin (110 μmol/l; normal range, 5.1–17.1
μmol/l), direct bilirubin (89 μmol/l; normal range,
0–6.0 μmol/l), alkaline phosphatase (1,005 U/l; normal
range, 30–110 U/l), γ-glutamyltranspeptidase (2,547 U/l; normal
range, 11–50 U/l) and lowered urinary specific gravity (1.0; normal
range, 1.005–1.030). The patient was negative for hepatitis viruses
A, B, C and E.
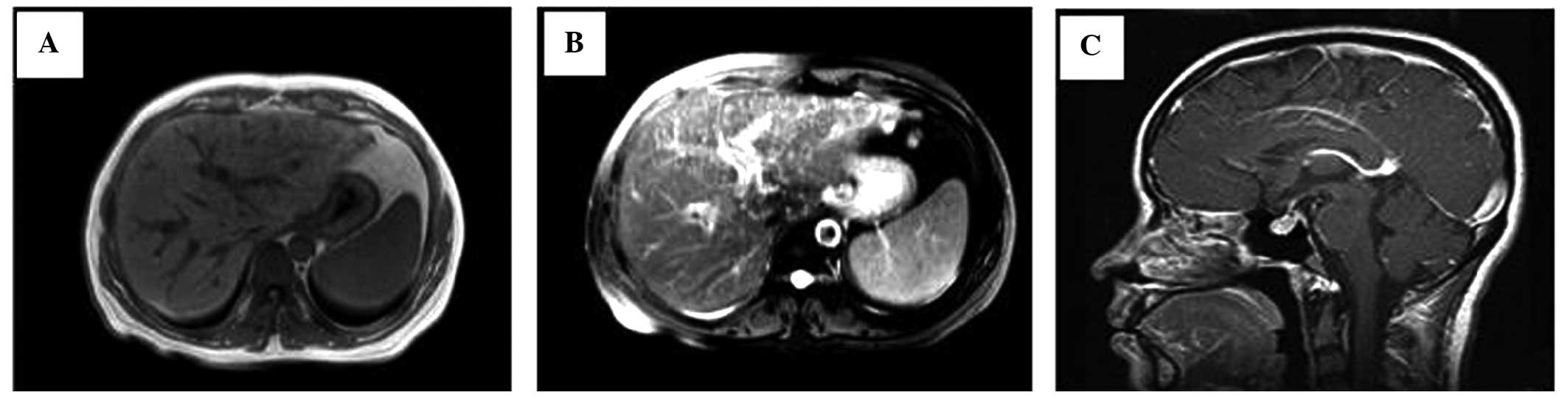
Abdominal ultrasound revealed enlarged spots on the
liver and splenomegaly. Abdominal MRI revealed splenomegaly and the
typical characteristics of diffuse liver cancer. For example, the
contrast-enhanced T1 signal revealed an uneven signal from the
liver parenchyma, miliary diffuse nodules and no expansion of the
intrahepatic bile duct (Fig. 1A).
Enhanced early-phase scanning indicated uneven perfusion,
arterial-phase scanning revealed significantly enhanced nodules,
and venous- and delayed-phase scans were marginally enhanced,
indicating the typical changes of liver cancer (Fig. 1B). Head MRI revealed an abnormal
skull shape with a thickened and enlarged pituitary stalk >3 mm
(Fig. 1C). The patient was
diagnosed with liver cancer and pituitary tumor.
However, liver histopathology revealed proliferation
of LCs, infiltration of eosinophils and other inflammatory cells,
including a number of foam cells and multinucleated giant cells
(Fig. 2A). Immunohistochemical
staining revealed CD1a-positive LCs (Fig. 2B). Therefore, the patient was
diagnosed with LCH, liver and spleen lesions, as well as central
diabetes insipidus.
The patient was treated with prednisolone (40
mg/m2/day), desmopressin (0.1 mg/m2/day) and
vincristine (6 mg/m2/week) for six weeks. During this
period, the symptoms of fever, fatigue and anorexia improved.
Volume of urine was reduced to 2,000–3,000 ml from an initial
6,000–8,000 ml every 24 h, but liver function tests revealed no
significant improvement. The initial therapy phase was extended for
another six weeks. During the following six weeks, oral
prednisolone (40 mg/m2/day) was administered three days
per week with vincristine (pushed 2 mg/m2/week). The
patient was followed-up for 10 months with treatment consisting of
prednisolone (40 mg/m2/day, days 1–5, Q3 weeks),
vinblastine (6 mg/m2/day, Q3 weeks) and 6-mercaptopurine
(MP) (50 mg/m2/day for seven months). As a result of the
treatment, liver function and blood cell tests improved.
Discussion
In the present case, liver damage was diagnosed by
skin and scleral jaundice as well as abnormal liver function tests.
Abdominal ultrasound and MRI revealed the typical changes of liver
cancer. The final diagnosis of LCH for this case mainly depended on
histological identification of LCH cells and CD1a-positive staining
of LCs. In addition, this particular case had three characteristics
that have rarely been reported previously: i) LCH occurred in an
adult with damage to three vital organs (the liver, spleen and
pituitary gland); ii) damage to the bone (which occurs in 92% of
cases) and skin (24% of cases), two of the most commonly affected
organs, was not detected; iii) a regimen of 12-week therapy of
prednisolone/desmopressin/vincristine plus maintenance therapy of
prednisolone/vinblastine/6-MP resulted in marked improvement in
symptoms and liver function (7).
LCH is a group of idiopathic disorders characterized
by the presence of cells with features similar to bone
marrow-derived LCs accompanied by a backdrop of hematopoietic
cells, including T-cells, macrophages and eosinophils. LCH occurs
in all age groups but usually affects children between one and 15
years old, with peak incidence between five and 10 years of age.
Liver images reveal nodular and irregular lesions, and spleen
images demonstrate splenomegaly (enlargement >2 cm below the
costal margin in the midclavicular line) (8). Radiological manifestations of the
disease include thickening of the pituitary stalk >3 mm, loss of
physiological hyperintense signal in the posterior pituitary on T1W
images, and loss of antidiuretic hormone storage granules (9). Diagnosis is confirmed histologically
by tissue biopsy. Appropriate histopathological tests are required
to establish a diagnosis of LCH. Heterogeneous admixture of cells
is also revealed by microscopy, including eosinophils,
polymorphonuclear leukocytes, giant cells and mononuclear cells, as
well as fibrosis. In cases of LCH, numerous mononuclear cells are
LCs, distributed diffusely or in clusters, which supports the
diagnosis (10). The
immunohistochemical confirmation of the presence of LCs by cell
surface CD1a antigen is useful for diagnosis. The CD1a surface
antigen can now be identified from routinely paraffin-embedded
specimens (11).
Thus far, the precise pathogenesis has not been
elucidated and there is no standard treatment for LCH. Currently,
there are four basic types of therapy for patients, including
debridement surgery, radiation therapy, chemotherapy and
immunotherapy, depending on the affected vital organs. However,
radiation therapy is only suitable for limited lesions, not for
multisystem disease. Due to the lack of a standard of care for the
treatment of LCH, three large-scale, international, prospective
therapeutic studies for multisystem LCH were conducted by the
Histiocyte Society in April, 2009 (6). LCH is divided into high- and low-risk
groups with different treatments for each. The low-risk group is
defined as patients older than two years without liver, bone marrow
or lung involvement. Patients with multiorgan involvement and
dysfunction of the liver, lungs or bone marrow, and lack of
response to initial therapy (assessed after 6–12 weeks of
treatment) are placed in the high-risk group. Therefore, the
patient reported in the present study fits the characteristics of
the high-risk group.
Different regimens for the treatment of adult LCH
with multisystem involvement have been reported with variable
efficacy (12). Natural mortality
of LCH is high, although patients have improved prognosis when
presenting with bone and skin lesions. Patients also have improved
prognosis should they respond to chemotherapy during the first six
weeks of treatment, regardless of whether there is multiple organ
damage. For the present case, a 12-week regimen of
prednisolone/desmopressin/vincristine was followed by maintenance
therapy of prednisolone/vinblastine/6-MP. Although this regimen
resulted in marked improvement in symptoms and liver function, the
patient did not respond well to the initial six-week therapy, and
no noticeable improvement in liver function was detected at first.
After 10 months of maintenance therapy, however, results of liver
function tests revealed significant improvement. To the best of our
knowledge, this represents a new treatment regimen. Therefore, this
study reports a rare case with multifocal disease and a unique
treatment. Although the patient was treated with a unique therapy
and survived for more than one year, further study is required.
References
|
1
|
Leonidas JC, Guelfguat M and Valderrama E:
Langerhans’ cell histiocytosis. Lancet. 361:1293–1295. 2003.
|
|
2
|
Gasent Blesa JM, Alberola Candel V, Solano
Vercet C, et al: Langerhans cell histiocytosis. Clin Transl Oncol.
10:688–696. 2008.
|
|
3
|
García Gallo MS, Martínez MP, Abalovich
MS, et al: Endocrine manifestations of Langerhans cell
histiocytosis diagnosed in adults. Pituitary. 13:298–303.
2010.PubMed/NCBI
|
|
4
|
Camelo-Piragua S, Zambrano E and
Pantanowitz L: Langerhans cell histiocytosis. Ear Nose Throat J.
89:112–113. 2010.
|
|
5
|
Howarth DM, Gilchrist GS, Mullan BP,
Wiseman GA, Edmonson JH and Schomberg PJ: Langerhans cell
histiocytosis: diagnosis, natural history, management, and outcome.
Cancer. 85:2278–2290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Minkov M, Grois N, McClain K, et al:
Langerhans Cell Histiocytosis. Histiocyte Society Evaluation and
Treatment Guidelines. 2009.
|
|
7
|
Kilpatrick SE, Wenger DE, Gilchrist GS, et
al: Langerhans’ cell histiocytosis (histiocytosis X) of bone. A
clinicopathologic analysis of 263 pediatric and adult cases.
Cancer. 76:2471–2484. 1995.
|
|
8
|
Mampaey S, Warson F, Van Hedent E and De
Schepper AM: Imaging findings in Langerhans’ cell histiocytosis of
the liver and the spleen in an adult. Eur Radiol. 9:96–98.
1999.
|
|
9
|
Redhu R, Nadkarni T and Mahesh R: Diabetes
insipidus associated with a thickened pituitary stalk in a case of
Langerhans cell histiocytosis. J Pediatr Neurosci. 6:62–64.
2011.
|
|
10
|
Lieberman PH, Jones CR, Steinman RM, et
al: Langerhans cell (eosinophilic) granulomatosis. A
clinicopathologic study encompassing 50 years. Am J Surg Pathol.
20:519–552. 1996.
|
|
11
|
Broadbent V, Gadner H, Komp DM and Ladisch
S: Histiocytosis syndromes in children: II. Approach to the
clinical and laboratory evaluation of children with Langerhans cell
histiocytosis Clinical Writing Group of the Histiocyte Society. Med
Pediatr Oncol. 17:492–495. 1989. View Article : Google Scholar
|
|
12
|
Minkov M, Grois N, Heitger A, et al:
Treatment of multisystem LCH. Results of DAL-HX 83 and DAL-HL 90
studies DAL-HX study group. Klin Padiatr. 212:139–144. 2000.
View Article : Google Scholar : PubMed/NCBI
|