Introduction
Excessive cell growth through the cell cycle is the
fundamental hallmark of cancer (1).
Cyclins and cyclin-dependent kinases (CDKs) drive the cell cycle
progression from G1 to S phase and G2 to M phase (2). Of the four CDKs (CDK1, CDK2, CDK4 and
CDK6), CDK4 and CDK6 are not required for the cell cycle of normal
cells but are essential for driving the cell cycle progression in
various types of cancer (3–5). It was previously reported that the
selective targeting of CDK4/6 kinase activity may block the cell
cycle and thus inhibit cancer growth (6). CDK4 and CDK6 interact with cyclin D
and form the cyclin D/CDK4 and cyclin D/CDK6 complexes where CDK4/6
are activated for G1-S transition through phosphorylation of the
retinoblastoma protein (RB) and its downstream E2F transcriptional
factors (7).
The development of small molecule inhibitors
targeting the cyclin-CDK4/6-RB axis for cancer therapy is crucial
(8). However, the first generation
of broad-range pan-CDK inhibitors such as flavopiridol (9) has not been of clinical benefit due to
the toxicity and lack of specificity (10). Efforts have been focused on the next
generation of CDK-specific inhibitors. PD-0332991 is a highly
selective, orally administered and reversible inhibitor of CDK4/6
(11,12). PD-0332991 blocks the cell cycle of
brain, breast, blood and pancreatic cancer cells in an RB-dependent
manner (13–17). Treatment of PD-0332991 inhibits the
growth of animal xenografts derived from these cancer cells
(11,15,18,19).
This selective CDK4/6 inhibitor is currently in phase I/II clinical
trials for advanced cancers and earlier data from the trials have
shown that PD-0332991 is well tolerated with a good safety profile
in cancer patients (20–22). Based on the preclinical and clinical
observations the potential of PD-0332991 for the treatment of
colorectal carcinoma was investigated.
Colorectal carcinoma is the third most common type
of cancer, but the second leading cause of cancer-related mortality
(23). Thus, development of novel
curative treatments for colorectal carcinoma is essential. The
cyclin D family includes cyclins D1, D2 and D3. Cyclin D1 is known
to be a predictive factor for therapeutic response of colorectal
carcinoma whereas cyclin D2 is required for the CDK4/6-driven
growth of colorectal adenoma cells (24). In addition, the E2F family protein
E2F4 is involved in the cell cycle progression of colorectal
carcinoma cells (25). Findings of
those studies suggest the possible role of cyclin D-CDK4/6-RB axis
in the growth of colorectal carcinoma. However, whether CDK4, CDK6
or both are required for the G1-S transition of colorectal
carcinoma cells remains to be clarified.
In this study, we showed that CDK6 and RB are highly
expressed in colorectal carcinoma tissues and derived cells as
compared to the matched normal colorectal tissues. Both CDK6 and RB
are required for the cell cycle progression of colorectal carcinoma
cells and by inhibiting the CDK6-RB axis, PD-0332991 induces the G1
cell cycle arrest and inhibits cancer cell growth. Thus, PD-0332991
may be used for the treatment of colorectal carcinoma.
Materials and methods
Human colorectal carcinoma and matched
normal tissues
Four human colorectal carcinoma and matched adjacent
normal colorectal tissue samples were collected in the Third
Hospital of Jilin University (Changchun, China) between January,
2010 and December, 2010, in accordance with the protocols approved
by the Institutional Review Board of the Third Hospital of Jilin
University. Patients provided written informed consent for the
tissue collection. This study was approved by the Third Hospital
Ethics Committee of Jilin University.
Human colorectal carcinoma cell
lines
The colorectal carcinoma cell lines CACO-2,
COLO-205, COLO-320, DLD-1, HCT-8, HT29 and SW948, together with the
human glioma cell line LN229 serving as the control, were purchased
from the American Type Culture Collection (ATCC, Manassas, VA,
USA). Each cell line was grown in RPMI-1640 medium (Invitrogen Life
Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (FBS) and maintained in a humidified 37°C and 5%
CO2 incubator (26).
Reagents and antibodies
PD-0332991 was purchased from Selleckchem (Houston,
TX, USA) and prepared as stock solutions in dimethyl sulfoxide
(DMSO). Antibodies against CDK1, CDK4, CDK6, cyclin D1, cyclin D3,
RB, phosphorylated-RB (pRB) (S780, S795 and S807/811) were
purchased from Cell Signaling Technology, Inc., (Danvers, MA, USA).
The antibody against CDK2 was purchased from Santa Cruz
Biotechnology, Inc., (Santa Cruz, CA, USA) and β-actin antibody was
purchased from Sigma-Aldrich (St. Louis, MO, USA). Horseradish
peroxidase-conjugated goat anti-mouse and goat anti-rabbit
antibodies were obtained from Jackson IR Laboratories, Inc., (West
Grove, PA, USA). Protease inhibitor mixture, Triton X-100 and other
chemicals were purchased from Sigma-Aldrich. The enhanced
chemiluminescence detection kit was obtained from Amersham
Biosciences (Piscataway, NJ, USA).
Western blotting
Western blotting was performed as previously
described (27). In brief, cell
lines and tissues were lysed in lysis buffer consisting of 20
mmol/l Tris pH 7.4, 150 mmol/l NaCl, 1% NP-40, 10% glycerol, 1
mmol/l EGTA, 1 mmol/l EDTA, 5 mmol/l sodium pyrophosphate, 50
mmol/l sodium fluoride, 10 mmol/l β-glycerophosphate, 1 mmol/l
sodium vanadate, 0.5 mmol/l DTT, 1 mmol/l PMSF, 2 mmol/l imidazole,
1.15 mmol/l sodium molybdate, 4 mmol/l sodium tartrate dihydrate
and 1X protease inhibitor cocktail (Sigma, St. Louis, MO, USA).
Following a 30-min incubation in lysis buffer at 4°C, lysates were
centrifuged at 18,000 × g for 15 min at 4°C. The supernatant was
collected and protein concentrations were determined by the
Bradford protein assay following the manufacturer’s instructions
(Bio-Rad, Hercules, CA, USA). Equal amounts of protein were
separated through SDS-PAGE gels and transferred onto nitrocellulose
membranes (Bio-Rad). The membranes were incubated overnight at 4°C
with primary antibody and then for 1 h with horseradish
peroxidase-conjugated secondary antibody. The membranes were
developed by chemiluminescence.
Lentiviral shRNA sequences and
transduction
Lentiviral shRNA vectors were purchased from the
Sigma MISSION® shRNA library (Sigma-Aldrich) and
included scrambled control (SHC002), CDK6-747 (TRCN0000039747,
5′-CGTGG AAGTTCAGATGTTGAT-3′) and CDK6-893 (TRCN000019 4893,
5′-CATGAGATGTTCCTATCTTAA-3′), CDK4-20 (TRCN0000010520,
5′-ACAGTTCGTGAGGTGGCTTTA-3′), and CDK4-64
(5′-ATGACTGGCCTCGAGATGTAC-3′). Each lentiviral shRNA vector was
transduced into cells that were selected with puromycin as
previously described (28).
Cell proliferation assay
Cell proliferation was determined by acid
phosphatase assay according to the manufacturer’s instructions
(29,30). In brief, untreated or transduced
cells with shRNA vectors were grown in 96-well plates at
1×103 cells per well in 200 μl of 10% FBS-containing
medium. Following incubation for 24 h, the medium was replaced with
10% FBS medium or the medium was supplemented with PD-0332991.
After incubation for 1, 3 or 5 days, the cells were washed with
phosphate-buffered saline (PBS) and each of the wells was added
with 100 μl buffer containing 0.2 M sodium acetate (pH 5.5), 0.2%
(v/v) Triton X-100 and 20 mmol/l p-nitrophenyl phosphate (Sigma 104
phosphatase substrate). The plates were incubated at 37°C for 1.5 h
and the reaction was stopped by the addition of 10 μl 1 M NaOH to
each well and staining was measured at 405 nm by a microplate
reader (Bio-Rad).
Flow cytometric analysis of the cell
cycle
Flow cytometry was performed as previously described
(26). In brief, cells were grown
in 65-mm plates at a density of 5×105 cells per well.
After 24 h incubation, the cells were grown for 24 h in 10% FBS
medium supplemented with or without PD-0332991 (1 μmol/l) in the
presence or absence of DMSO as a control. After treatment, cells
were collected, washed with PBS and fixed by incubation in 70%
ethanol solution at 4°C. The fixed cells were washed and the cell
pellets were stained using a propidium iodide-RNase solution (PBS
containing 20 μg/ml propium iodide, 20 μg/ml DNase-free RNase A and
0.1% Triton X-100) for 30 min at 20°C in the dark. The cell cycle
status was analyzed with a flow cytometer using FlowJo software
(Tree Star, Inc., Ashland, OR, USA).
Statistical analysis
Data were presented as the means ± standard
deviation (SD) and analyzed statistically by Student’s t-test.
P<0.05 was considered statistically significant.
Results
CDK proteins are overexpressed in human
colorectal carcinoma
To the best of our knowledge, this is the first
study concerning the expression of cell cycle proteins in
colorectal carcinoma tissues. We examined the expression of key
cell cycle proteins in surgically resected colorectal carcinoma
tissues as compared with matched adjacent normal colorectal
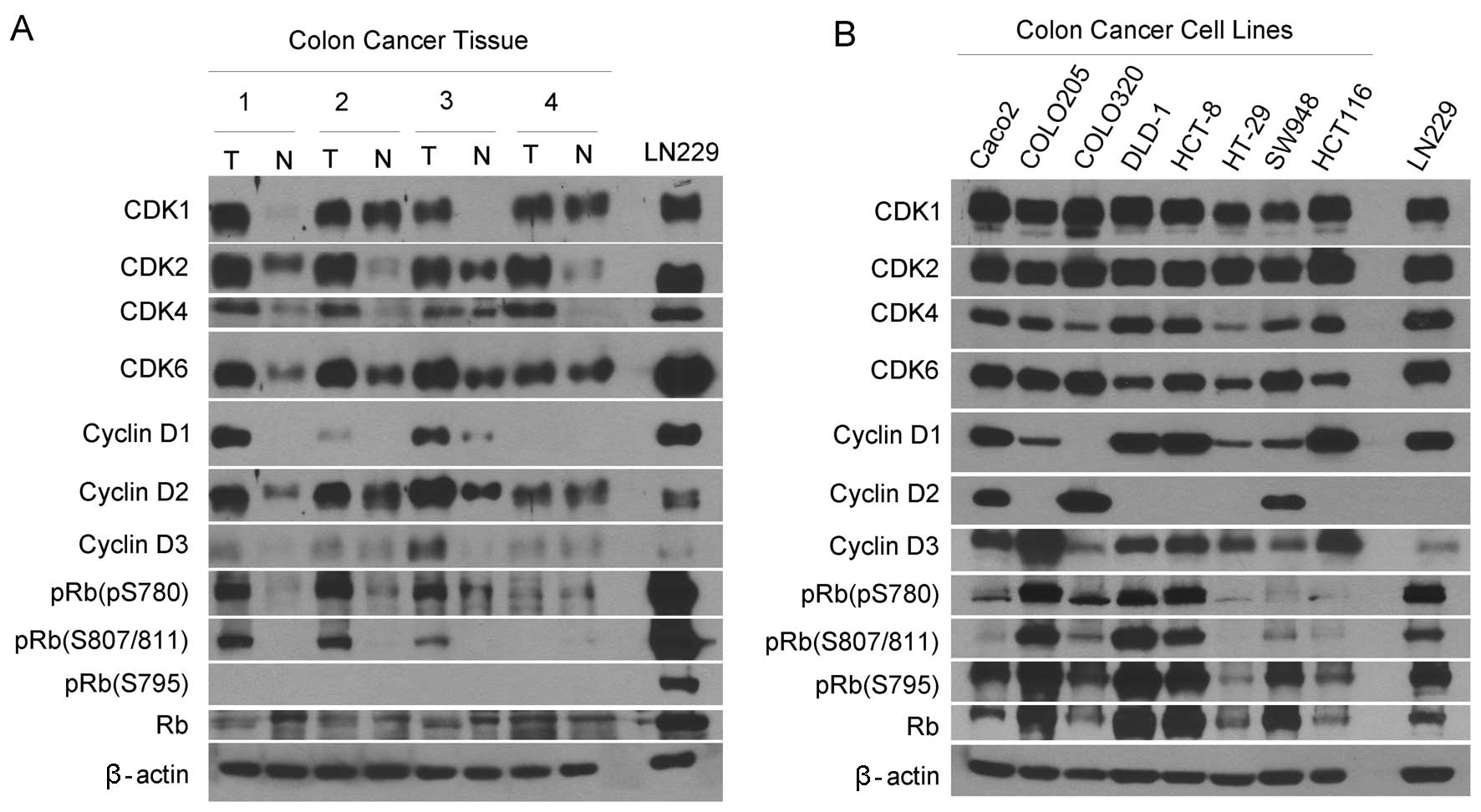
tissues. Western blotting revealed that CDK1, CDK2, CDK4 and CDK6
proteins were expressed at much higher levels in the carcinoma
tissues than the matched normal tissues (Fig. 1A). These CDK proteins were expressed
in the carcinoma-derived cell lines (Fig. 1B). Cyclin D1 was detected in half of
the carcinoma tissues and cyclin D3 was observed in only one of the
four carcinoma tissues, while cyclin D1 and D3 were slightly
detected in normal tissues. By contrast, cyclin D2 was expressed in
all the carcinoma and matched normal tissues with the expression
levels being higher in the carcinoma tissues. Consistent with this
profile, cyclin D1 was highly expressed in four but weakly in
three; cyclin D2 was highly expressed in three and cyclin D3 was
expressed in seven of eight cell lines.
We examined the expression of unphosphorylated RB
and pRB. RB can be phosphorylated at several serine residues
including S780, S795, S807 and S811 (31). Thus, the antibodies against pRB
(S780), pRB (S795) and pRB (S808/811) were used in this study.
Western blotting detected pRB (S780) and pRB (S808/811) in three of
four carcinoma tissues but only slightly in the normal tissues
(Fig. 1A). The pRB (S795) was
observed only in the cell lines but not any tissues as compared
with the expression in the control glioblastoma cell line LN229. By
contrast, pRB (S795) was detected in all the cell lines (Fig. 1B). Collectively, the G1 cyclins, CDK
and pRB are expressed differentially in human colorectal carcinoma
tissues and cell lines.
PD-0332991 treatment induces G1 arrest in
colorectal carcinoma cells
To explore the potential of PD-0332991 in treating
colorectal carcinoma, we examined whether the carcinoma cells
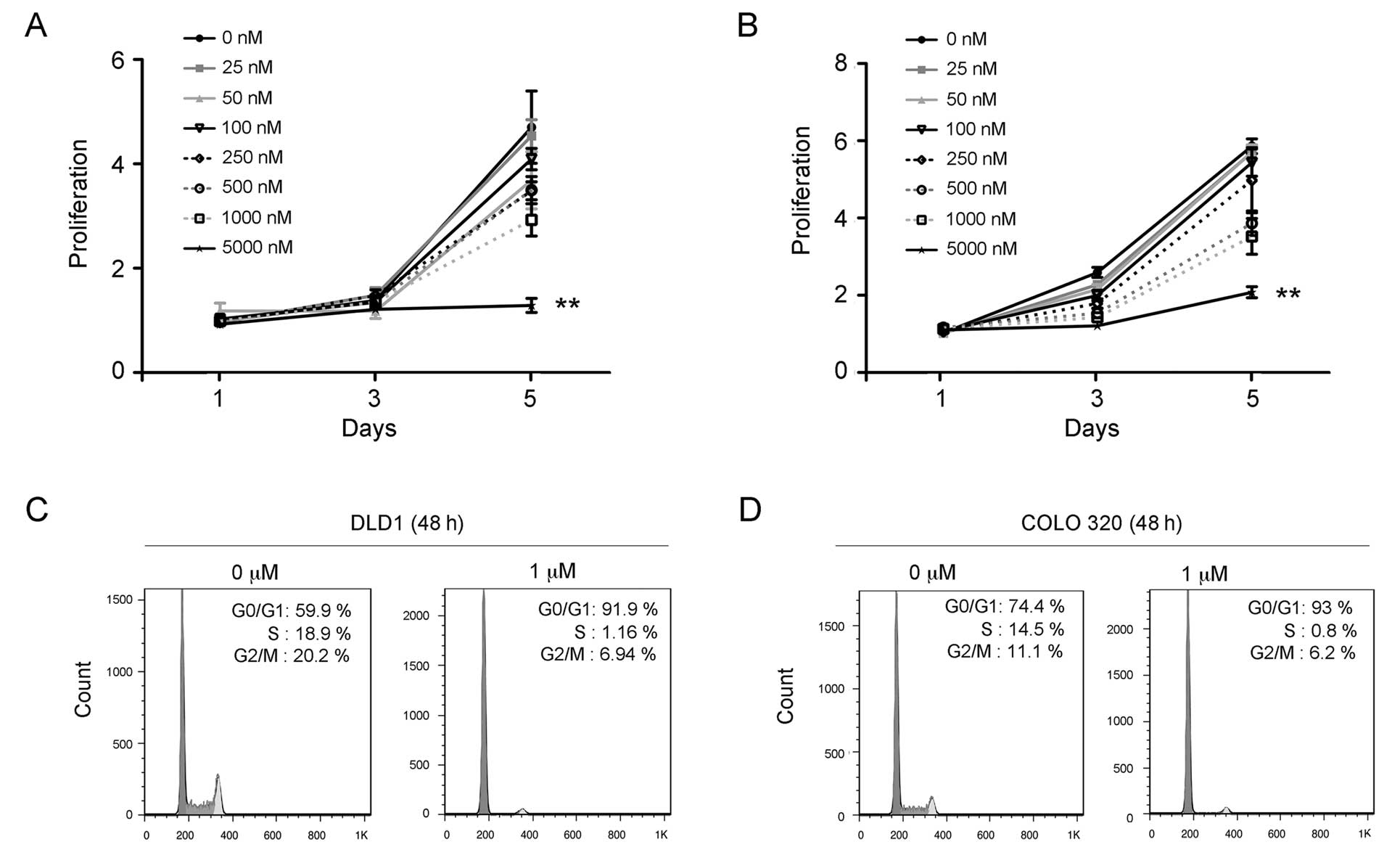
respond to PD-0332991 treatment in culture. The carcinoma cell
lines DLD-1 and COLO320 were treated with PD-0332991 concentrations
ranging between 25 nM and 5000 nM for 72 h. Results of the cell
proliferation assay showed that PD-0332991 treatment significantly
inhibited the growth of DLD-1 (Fig.
2A) and COLO320 cells in a dose-dependent manner (Fig. 2B). To evaluate whether inhibition by
PD-0332991 occurs through the cell cycle, DLD-1 and COLO320 cells
were treated or untreated with PD-0332991 (1 μM) for 48 h and
subjected to flow cytometry for the cell cycle as previously
described (30). The results showed
that the PD-0332991 treatment led to a significant increase in the
number of G1 phase cells but a marked decrease of the S phase cells
in the DLD-1 (Fig. 2C) and COLO320
cells (Fig. 2D). These findings
suggest that PD-0332991 treatment inhibits growth through the
induction of G1 arrest of colorectal carcinoma cells.
PD-0332991 treatment inhibits RB
phosphorylation in colorectal carcinoma cells
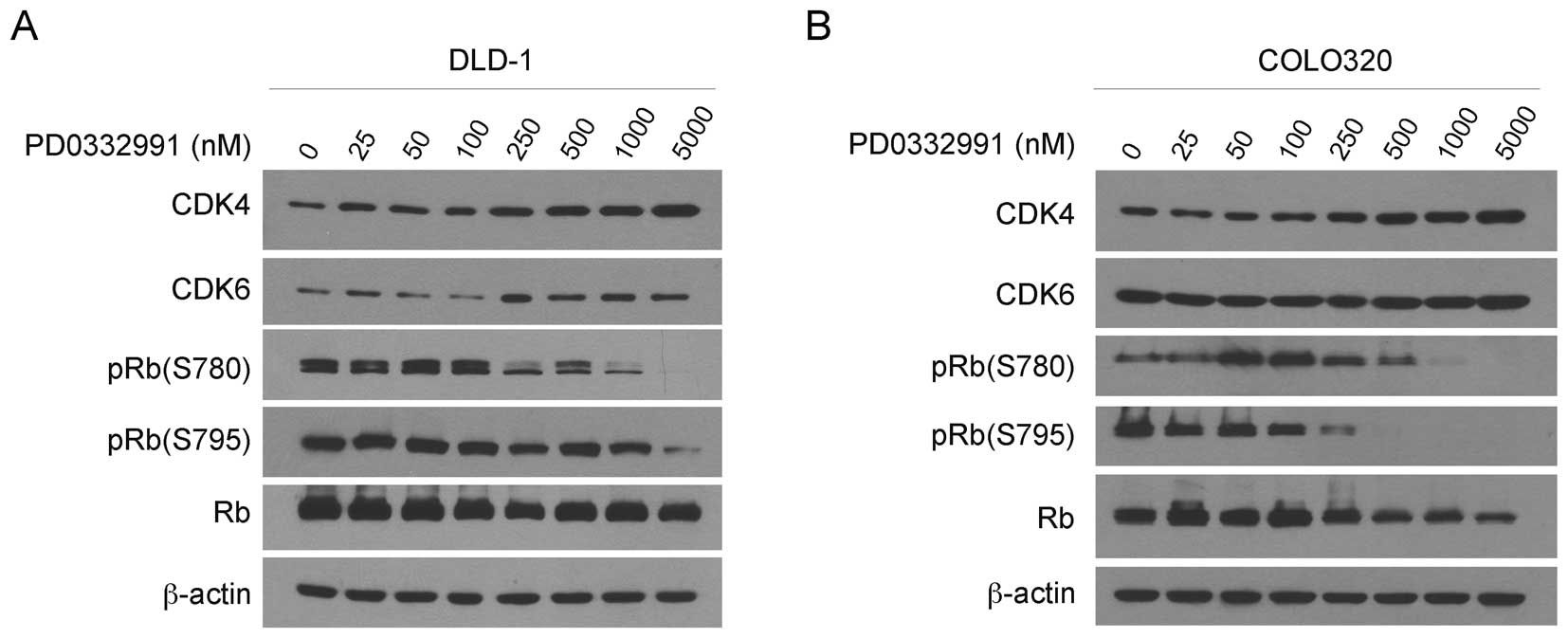
To examine the mechanisms in the PD-0332991-induced
G1 arrest, we examined the CDK4/6 and RB proteins in DLD-1 and
COLO320 cells after 24 h treatment with a series of concentrations
of PD-0332991. Western blotting revealed that the treatment did not
affect the levels of CDK4, CDK6 and unphosphorylated RB in DLD-1
(Fig. 3A) and COLO320 cells
(Fig. 3B). By contrast, PD-0332991
treatment markedly reduced the levels of pRB (S780) and pRB (S795)
in each of the cell lines in a dose-dependent manner. Collectively,
PD-0332991 treatment inhibits RB phosphorylation, induces G1 arrest
and thus suppresses the growth of colorectal carcinoma cells in
culture.
CDK6 phosphorylates RB for the growth of
colorectal carcinoma cells
CDK4 and CDK6 regulate the G1-S cell cycle
transition (2). However, whether
CDK4 or CDK6 or both were required for RB phosphorylation and G1-S
transition in colorectal carcinoma cells remained to be clarified.
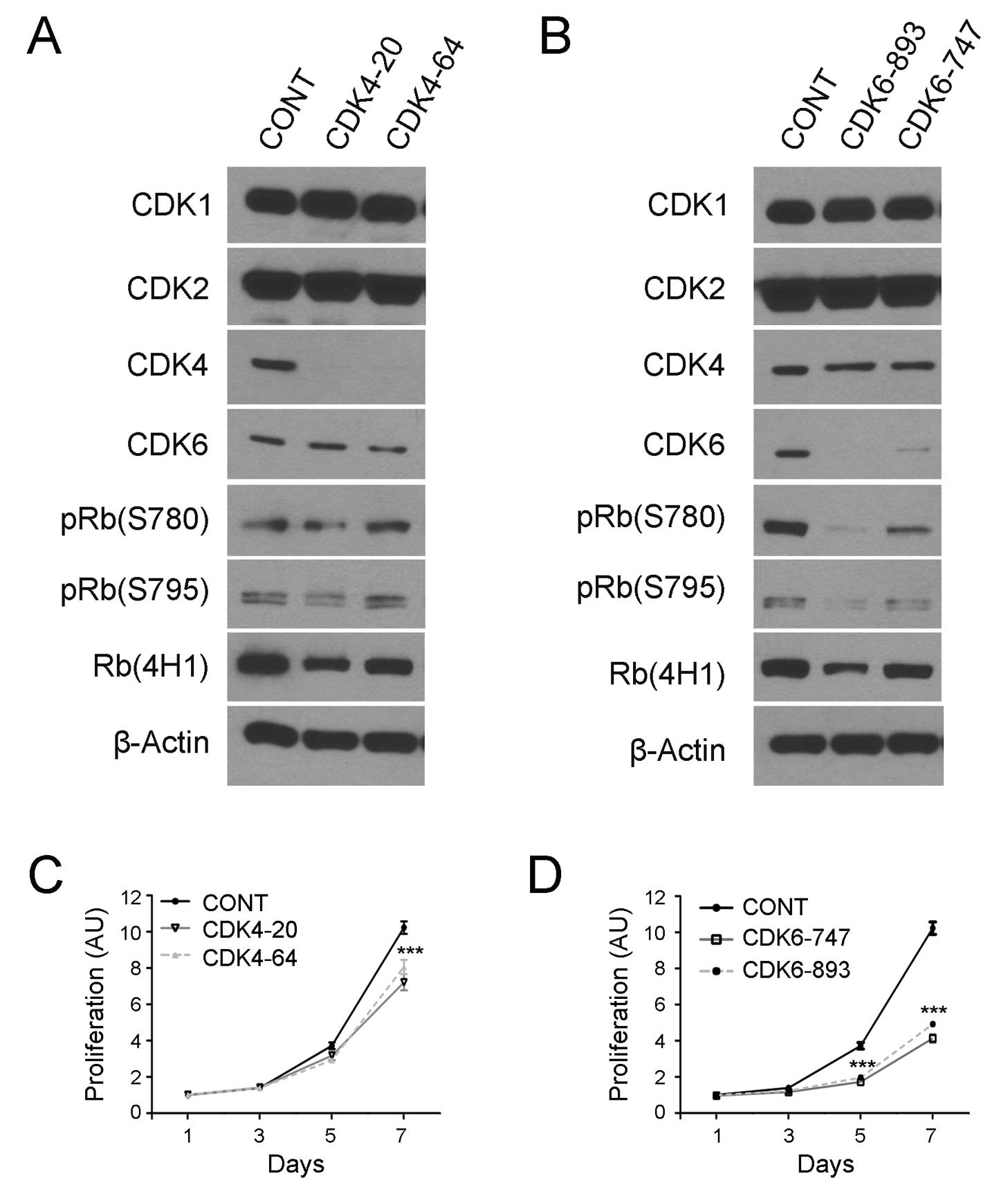
To address this issue, lentiviral vectors encoding
CDK4/CDK6-specific shRNA sequences were transduced into COLO320
cells as previously described (28). To prevent off-target effects, two
shRNA target sequences were used for each of the kinases including
CDK4-20 and CDK4-64 to target CDK4 and CDK6-747 and CDK6-893 for
CDK6 knockdown. The transduced cells were examined by western
blotting and the results revealed that the transduction of the CDK4
and CDK6 shRNA encoding vectors eliminated the expression of CDK4
(Fig. 4A) and CDK6 protein
(Fig. 4B), respectively, in COLO320
cells.
Notably, CDK4 knockdown did not affect RB
phosphorylation as evidenced by western blotting using the pRB
(S780) and pRB (S795) antibodies (Fig.
4A). By contrast, CDK6 knockdown markedly reduced the
expression of these phosphorylated RB proteins in the carcinoma
cells (Fig. 4B). To examine the
effects of CDK4/6 knockdown and RB phosphorylation inhibition on
cancer cell growth, we transduced the shRNA-coded vectors in
COLO320 cells, selected stably transduced cells and examined cell
growth using a cell viability assay. The results showed that the
knockdown of CDK4 slightly inhibited the growth of COLO320 cells
(Fig. 4C), whereas the knockdown of
CDK6 resulted in a marked inhibition of the growth of COLO320 cells
(Fig. 4D). The data suggest that
CDK6 plays a critical role in RB phosphorylation and cell growth in
colorectal carcinoma cells.
Discussion
Colorectal carcinoma is the second leading cause of
cancer-related mortality due to the lack of curative treatments
(23). Previously, signaling
pathways were found to be involved in the formation and progression
of colorectal carcinoma for the development of cancer signal
pathway-targeted therapies. However, such targeted therapies have
not been materialized for the effective treatment of this lethal
cancer (32). Novel therapeutic
agents are therefore required for treatment of this type of cancer.
In the present study, the therapeutic potential of the selective
CDK4/6 inhibitor PD-0332991 in treating colorectal carcinoma was
demonstrated.
The cyclin D-CDK4 and cyclin-CDK6 complex drives the
cell cycle through G1-S transition via phosphorylation of RB in
various types of cancer (2). Thus,
therapeutic agents have been generated targeting these G1 phase
kinases for cancer therapies (6).
Of these novel therapeutic agents, the CDK4/CDK6 selective and
potent inhibitor PD-0332991 (11)
has passed the safety test with anticancer activity in phase I/II
trials (20–22). However, there are currently no
studies regarding the therapeutic potential of this inhibitor in
the treatment of colorectal carcinoma. Similarly, there are few
studies with regard to the cell cycle pathway in this cancer.
The data presented herein have shown that the G1
phase cyclin D1, D2, D3, CDK4, CDK6 and pRB proteins are highly
expressed in colorectal carcinoma tissues as compared to the
matched adjacent normal colorectal tissues. CDK4 and CDK6 control
the G1-S transition through the cell cycle (2) and recent studies of
genetically-engineered mice suggest that CDK4 or CDK6 is used to
drive the cell cycle in each type of cancer (3–5). To
identify the G1 kinase in colorectal carcinoma, we have shown that
knockdown of CDK6 but not CDK4 markedly reduces RB phosphorylation
and inhibits the growth of colorectal carcinoma cells, suggesting
for the first time that CDK6-RB axis is crucial in the growth of
the carcinoma and targeting of the CDK6-RB axis may provide a novel
therapeutic strategy in the treatment of colorectal carcinoma.
Preclinical studies have demonstrated the anticancer
activities of PD-0332991 in treating brain, breast, blood and
pancreatic cancers in an RB-dependent manner (13–17).
To the best of our knowledge, this study has shown for the first
time that PD-0332991 treatment blocks RB phosphorylation, induces
G1 arrest and thus inhibits the growth of human colorectal
carcinoma cells. This study therefore suggests that PD-0332991 has
therapeutic effects against colorectal carcinomas through
inhibition of the CDK6/RB pathway. Thus, cell cycle pathways in
colorectal carcinoma and the development of PD-0332991 into an
effective therapeutic agent for clinical treatment of human
colorectal carcinoma should be investigated.
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar
|
|
2
|
Malumbres M: Cell cycle-based therapies
move forward. Cancer Cell. 22:419–420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu Q, Sicinska E, Geng Y, et al:
Requirement for CDK4 kinase function in breast cancer. Cancer Cell.
9:23–32. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Puyol M, Martin A, Dubus P, et al: A
synthetic lethal interaction between K-Ras oncogenes and Cdk4
unveils a therapeutic strategy for non-small cell lung carcinoma.
Cancer Cell. 18:63–73. 2010. View Article : Google Scholar
|
|
5
|
Hu MG, Deshpande A, Enos M, et al: A
requirement for cyclin-dependent kinase 6 in thymocyte development
and tumorigenesis. Cancer Res. 69:810–818. 2009. View Article : Google Scholar
|
|
6
|
Blagden S and de Bono J: Drugging cell
cycle kinases in cancer therapy. Curr Drug Targets. 6:325–335.
2005. View Article : Google Scholar
|
|
7
|
Malumbres M, Sotillo R, Santamaria D, et
al: Mammalian cells cycle without the D-type cyclin-dependent
kinases Cdk4 and Cdk6. Cell. 118:493–504. 2004. View Article : Google Scholar
|
|
8
|
Collins I and Garrett MD: Targeting the
cell division cycle in cancer: CDK and cell cycle checkpoint kinase
inhibitors. Curr Opin Pharmacol. 5:366–373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu G, Gandara DR, Lara PN Jr, et al: A
Phase II trial of flavopiridol (NSC #649890) in patients with
previously untreated metastatic androgen-independent prostate
cancer. Clin Cancer Res. 10:924–928. 2004.
|
|
10
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fry DW, Harvey PJ, Keller PR, et al:
Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991
and associated antitumor activity in human tumor xenografts. Mol
Cancer Ther. 3:1427–1438. 2004.
|
|
12
|
Toogood PL, Harvey PJ, Repine JT, et al:
Discovery of a potent and selective inhibitor of cyclin-dependent
kinase 4/6. J Med Chem. 48:2388–2406. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wiedemeyer WR, Dunn IF, Quayle SN, et al:
Pattern of retinoblastoma pathway inactivation dictates response to
CDK4/6 inhibition in GBM. Proc Natl Acad Sci USA. 107:11501–11506.
2010. View Article : Google Scholar
|
|
14
|
Finn RS, Dering J, Conklin D, et al: PD
0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially
inhibits proliferation of luminal estrogen receptor-positive human
breast cancer cell lines in vitro. Breast Cancer Res. 11:R772009.
View Article : Google Scholar
|
|
15
|
Michaud K, Solomon DA, Oermann E, et al:
Pharmacologic inhibition of cyclin-dependent kinases 4 and 6
arrests the growth of glioblastoma multiforme intracranial
xenografts. Cancer Res. 70:3228–3238. 2010. View Article : Google Scholar
|
|
16
|
Baughn LB, Di Liberto M, Wu K, et al: A
novel orally active small molecule potently induces G1 arrest in
primary myeloma cells and prevents tumor growth by specific
inhibition of cyclin-dependent kinase 4/6. Cancer Res.
66:7661–7667. 2006. View Article : Google Scholar
|
|
17
|
Liu F and Korc M: Cdk4/6 inhibition
induces epithelial-mesenchymal transition and enhances invasiveness
in pancreatic cancer cells. Mol Cancer Ther. 11:2138–2148. 2012.
View Article : Google Scholar
|
|
18
|
Cen L, Carlson BL, Schroeder MA, et al:
p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase
inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol.
14:870–881. 2012. View Article : Google Scholar
|
|
19
|
Roberts PJ, Bisi JE, Strum JC, et al:
Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer
therapy. J Natl Cancer Inst. 104:476–487. 2012. View Article : Google Scholar
|
|
20
|
Schwartz GK, LoRusso PM, Dickson MA, et
al: Phase I study of PD 0332991, a cyclin-dependent kinase
inhibitor, administered in 3-week cycles (Schedule 2/1). Br J
Cancer. 104:1862–1868. 2011. View Article : Google Scholar
|
|
21
|
Flaherty KT, Lorusso PM, Demichele A, et
al: Phase I, dose-escalation trial of the oral cyclin-dependent
kinase 4/6 inhibitor PD 0332991, administered using a 21-day
schedule in patients with advanced cancer. Clin Cancer Res.
18:568–576. 2012. View Article : Google Scholar
|
|
22
|
Leonard JP, LaCasce AS, Smith MR, et al:
Selective CDK4/6 inhibition with tumor responses by PD0332991 in
patients with mantle cell lymphoma. Blood. 119:4597–4607. 2012.
View Article : Google Scholar
|
|
23
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
24
|
Cole AM, Myant K, Reed KR, et al: Cyclin
D2-cyclin-dependent kinase 4/6 is required for efficient
proliferation and tumorigenesis following Apc loss. Cancer Res.
70:8149–8158. 2010. View Article : Google Scholar
|
|
25
|
Garneau H, Paquin MC, Carrier JC and
Rivard N: E2F4 expression is required for cell cycle progression of
normal intestinal crypt cells and colorectal cancer cells. J Cell
Physiol. 221:350–358. 2009. View Article : Google Scholar
|
|
26
|
Li B, Gao S, Wei F, Bellail AC, Hao C and
Liu T: Simultaneous targeting of EGFR and mTOR inhibits the growth
of colorectal carcinoma cells. Oncol Rep. 28:15–20. 2012.
|
|
27
|
Wei F, Liu Y, Bellail AC, et al: K-Ras
mutation-mediated IGF-1-induced feedback ERK activation contributes
to the rapalog resistance in pancreatic ductal adenocarcinomas.
Cancer Lett. 322:58–69. 2012. View Article : Google Scholar
|
|
28
|
Bellail AC, Olson JJ, Yang X, Chen ZJ and
Hao C: A20 ubiquitin ligase-mediated polyubiquitination of RIP1
inhibits caspase-8 cleavage and TRAIL-induced apoptosis in
glioblastoma. Cancer Discov. 2:140–155. 2012. View Article : Google Scholar
|
|
29
|
Wang Q, Wei F, Li C, et al: Combination of
mTOR and EGFR kinase inhibitors blocks mTORC1 and mTORC2 kinase
activity and suppresses the progression of colorectal carcinoma.
PLoS One. 8:e731752013. View Article : Google Scholar
|
|
30
|
Wang Q, Wei F, Lv G, et al: The
association of TP53 mutations with the resistance of colorectal
carcinoma to the insulin-like growth factor-1 receptor inhibitor
picropodophyllin. BMC Cancer. 13:5212013. View Article : Google Scholar
|
|
31
|
Rubin SM: Deciphering the retinoblastoma
protein phosphorylation code. Trends Biochem Sci. 38:12–19. 2013.
View Article : Google Scholar
|
|
32
|
Meyerhardt JA and Mayer RJ: Systemic
therapy for colorectal cancer. N Engl J Med. 352:476–487. 2005.
View Article : Google Scholar : PubMed/NCBI
|