Introduction
Primitive neuroectodermal tumors (PNETs) were first
discovered by Hart and Earle in 1973 (1). PNETs are small, round cell tumors that
undergo neuroectodermal differentiation. PNETs exhibit chromosomal
translocations in common with those of Ewing’s sarcoma (EWS) and
are thus often called Ewing family tumors (EFTs). PNETs are
classified as central or peripheral types based on their site of
origin, they usually originate in bone or soft tissue, but rarely
arise in the vulva. A diagnosis normally based on morphological
features and the immunohistochemical staining profile in addition
to cytogenesis and clinical symptoms. Typical histological features
include small, uniform sized round cells with hyperchromatic nuclei
and scant cytoplasms. The tumors exhibit neural differentiation by
forming a rosette-like structure. However, they are normally
diagnosed by immunohistochemical methods. In the vast majority of
cases, PNETs have been shown to express at extremely high levels,
an antigen determined by the MIC2 gene. PNET patients usually
experience pain as the tumors may develop in almost any bone or
soft tissue. Approximately 25% of patients have detectable
metastatic lesions in the lung, bone and bone marrow at diagnosis.
The current case report presents a case of PNET originating in the
vulva.
Case report
Patient presentation
A 60-year-old female visited Osaka City Sumiyoshi
Hospital (Osaka, Japan) for evaluation of a mass in the right side
of the vulva. Enucleation of the vulvar tumor was performed and the
suspected diagnosis was hemangiopericytoma. The tumor exhibited
characteristics of borderline malignancy and the patient was
referred to Osaka City University Hospital (Osaka, Japan) for
additional therapy. Informed consent was obtained from the patient
for the use of clinical information for education and research.
The tissue excised at the previous hospital was
histopathologically examined by the pathologist and the suspected
diagnosis was PNET. There were no abnormal findings in the physical
examination. Magnetic resonance imaging (MRI) and computed
tomography did not reveal any residual tumor between the lungs and
the vulva.
Simple vulvectomy and resection of the inguinal
lymph nodes were performed, showing no microscopic residual tumor
presence. The patient underwent outpatient observation.
Four years following the initial surgery, a mass in
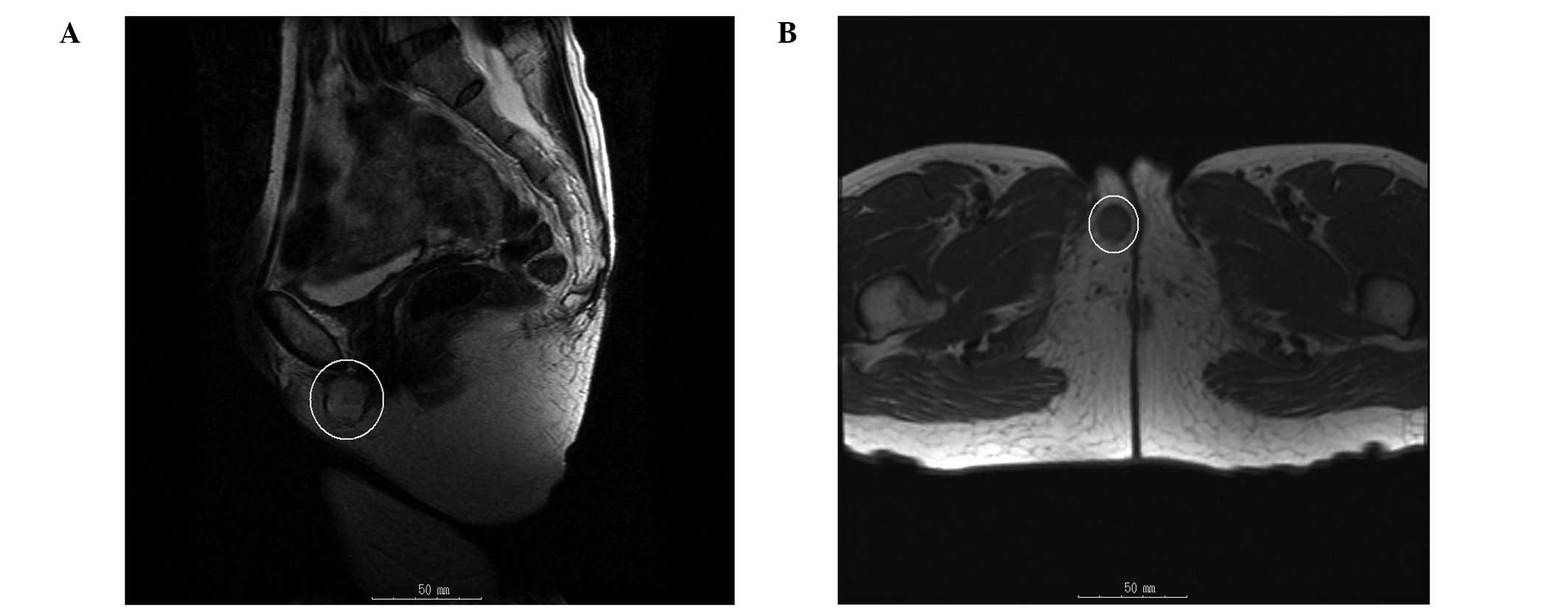
the right side of the vulva was observed. MRI imaging revealed a
3-cm mass in the right side of the vulva. T1-weighted images
demonstrated low signal intensity and T2-weighted images
demonstrated high signal intensity (Fig. 1). The possibility of recurrence was
suspected and the tumor was excised. The excised tumor was a
yellow-white, solid, soft and elastic mass.
Pathology
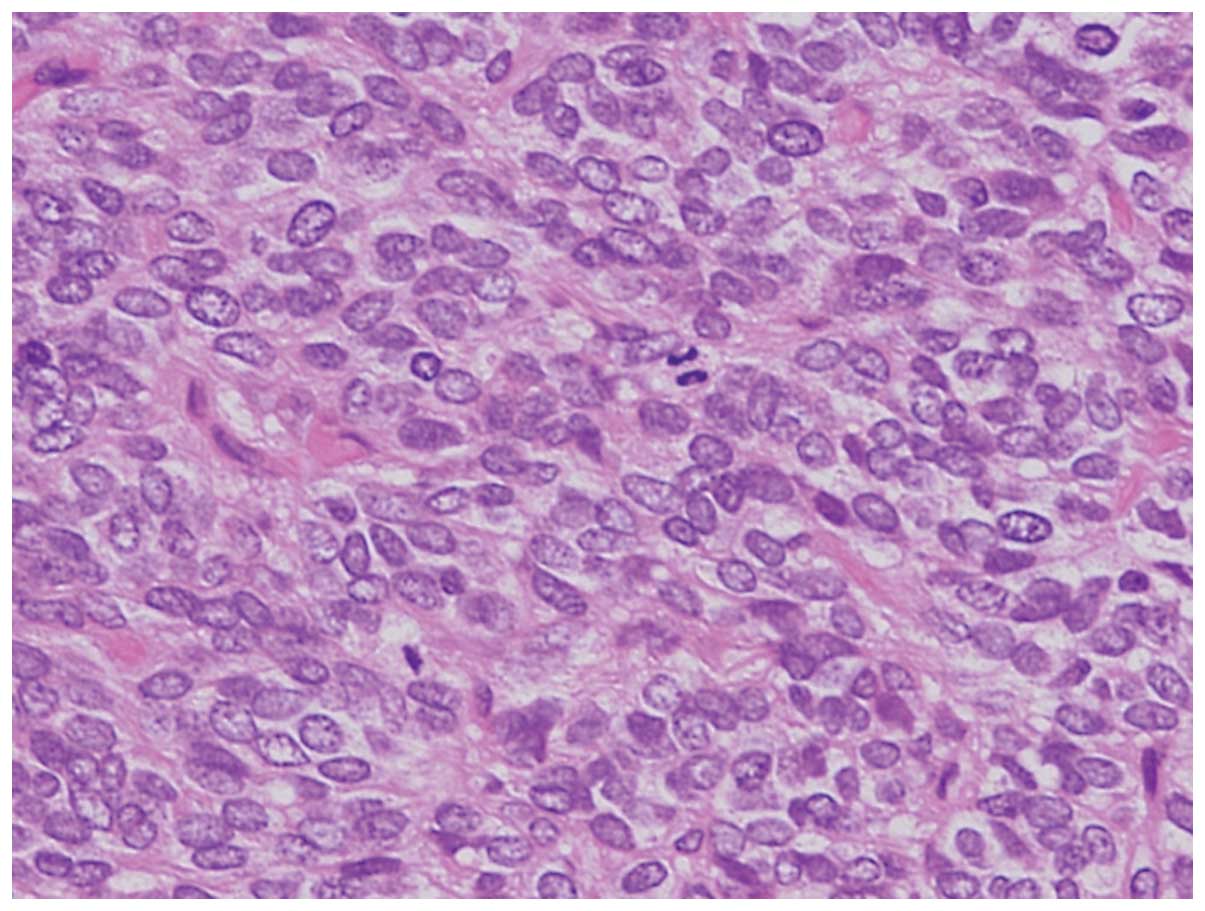
Hematoxylin and eosin-stained sections of the tumor
showed a solid growth pattern. The tumor comprised small,
round-to-oval nuclei. Homer-Wright rosettes were not observed
(Fig. 2).
Immunohistochemistry
The tumor stained positively for MIC-2,
synaptophysin, neuron-specific enolase (NSE) and neurofilament
antibodies. The tumor cells were negative for periodic-acid Schiff,
vimentin, desmin, chromogranin A, CD34, CD45 [leukocyte common
antigen (LCA)], S100, α-smooth muscle actin (α-SMA) and CD117
antibodies (Table I).
 | Table IImmunohistochemistry results. |
Table I
Immunohistochemistry results.
| Antibody | Staining result |
|---|
| MIC-2 | + |
| Synaptophysin | + |
| NSE | + |
| Neurofilament | + |
| Chromogranin | − |
| PAS | − |
| Vimentin | − |
| Desmin | − |
| Chromogranin A | − |
| CD34 | − |
| CD45 | − |
| S100 | − |
| αSMA | − |
| CD117 | − |
Clinical course
The patient was diagnosed with recurrent PNET that
had originated in the vulva. Multidisciplinary therapy was
administered. Combination chemotherapy, using vincristine,
tetrahydropyranyl-adriamycin and cyclophosphamide (VAC), was
administered and radiation therapy, ifosfamide chemotherapy and VAC
therapy were subsequently performed. The only adverse event
observed with this medical treatment was G3 myelosuppression.
Following multidisciplinary therapy, the patient underwent further
outpatient observation. To date, the patient remains alive with no
evidence of disease four years following the final medical
treatment.
Discussion
PNETs were first reported in 1918 by Stout (2) as small round cell tumors arising in
the ulnar bone with rosette formation. Hart and Earle (1) described the term PNET in 1973. PNETs
are classified as central or peripheral types based on their site
of origin. Peripheral PNETs usually originate in the bone or soft
tissue and often from genital organs (3,4).
However, a small number of reports have described PNETs originating
in the vulva. Fewer than 20 case reports were found in a search of
current literature (5–9).
Other small round cell tumor types include
neuroblastoma, malignant lymphoma and rhabdomyoblastoma. The tumor
in the present case was immunohistologically positive for MIC-2
and, therefore, the patient was diagnosed with an EFT. Notably,
markers of nervous system involvement, including NSE,
neurofilaments and synaptophysin, were also positive. The patient
was therefore diagnosed with recurrent PNET that originated in the
vulva. In the present case, neuroblastoma was excluded, as the
tumor was immunohistochemically positive for MIC-2, and malignant
lymphoma was excluded, as the tumor was immunohistochemically
negative for LCA. Rhabdomyoblastoma was also excluded, due to a
negative result for desmin and α-SMA. However, the
immunohistochemistry of PNETs is controversial and identification
of the chromosomal translocations is effective in determining the
diagnosis. The chimeric chromosomes associated with EWS/PNETs are
EWS-friend leukemia virus integration 1 (FLI1) (11;22)(q24;q12),
EWS-ETS-related gene (ERG) (21;22)(q22;q12), EWS-ETS variant 1
(7;22)(q22;q12), EWS-EIAF (17;22)(q121;q12) and EWS-FEV
(2;22)(q33;q12). All chimeric chromosomes exhibit EWS at the 5′-end
and transcription factors of the ETS family, associated with cell
proliferation, at the 3′-end. They are classified as EWS/ETS
chimeric chromosomes due to this feature. EWS is present on
chromosome 22 and gene expression of EWS is observed in various
organs. EWS/ETS chimeric chromosomes take part in cell
proliferation, through transcription or transformation activities,
and affect cultured fibroblasts, thereby promoting neural and
epithelial differentiation (10).
Overall, >90% of EWS/PNETs contain chimeric chromosomes of
EWS-FLI1 and EWS-ERG, in particular EWS-FLI1 translocation, which
is present in >80% of EWS/PNETs (11,12).
Poor prognostic factors in EWS/PNETs include lesions
of the body trunk, patient ages of ≥15 years, tumor volumes of ≥200
ml, the presence of distant metastasis, high levels of serum
lactate dehydrogenase, a poor response to chemotherapy and
recurrence within two years. The most unfavorable prognostic factor
is the presence of distant metastasis. Even with aggressive
treatment, patients with metastases exhibit an ~20% chance of
long-term survival (13). Despite
having no distant metastatic tumors, the patient in the current
case report exhibited unfavorable prognostic factors with regard to
the location of the lesion (body trunk) and the age of the
individual (60 years).
There are several reports on the management of
PNETs. The majority of PNETs are treated as EFTs. Multidisciplinary
therapy is often performed for EFTs as recurrence is common. Prior
to the advent of modern chemotherapy, <10% of patients with EFT
survived beyond five years following diagnosis (14). The Intergroup Ewing’s Sarcoma Study
and Cooperative Ewing Sarcoma Study recommend the standard
chemotherapy comprising between four and six drugs, including
doxorubicin, cyclophosphamide, vincristine, ifosfamide, etoposide
and actinomycin (15). However, a
standard therapy for recurrent tumors has not been established and
autologous peripheral blood stem cell transfusion following
chemotherapy is occasionally reported (16). In one study, the five-year survival
rate for patients with recurrence at local sites was 31.4±11.6% in
the surgical group and 9.1±6.1% in the non-surgical group (17). The effectiveness of the
aforementioned chemotherapy or surgery protocols for older
patients, as in the present case, remains unclear, due to the
majority of studies on the treatment of EFT involving young
patients.
The current case report presented a rare case of
PNET originating in the vulva. Although PNETs usually exhibit a
poor prognosis, the patient remains alive and with no evidence of
disease. It is hypothesized that this may be due to the 3-cm tumor
size and the absence of distant metastasis at the time of
recurrence.
References
|
1
|
Hart NH and Earle KM: Primitive
neuroectodermal tumors of the brain in children. Cancer.
32:890–897. 1973.
|
|
2
|
Stout AP: A tumor of the ulnar nerve. Proc
NY Pathol Soc. 18:2–12. 1918.
|
|
3
|
Chen L: Primitive neuro ectodermal tumor
of the uterus. Arch Histopathol D D. 16:33–36. 2009.
|
|
4
|
Fischer G, Odunsi K, Lele S and Mhawech P:
Ovarian primary primitive neuroectodermal tumor coexisting with
endometrioid adenocarcinoma: a case report. Int J Gynecol Pathol.
25:151–154. 2006.
|
|
5
|
Halil S, Kucuk M, Arvas M, et al:
Peripheral primitive neuroectodermal tumor (PNET) of the vulva: a
case report. Eur J Gynaecol Oncol. 32:117–118. 2011.
|
|
6
|
Scherr GR, d’Ablaing G III and Ouzounian
JG: Peripheral primitive neuroectodermal tumor of the vulva.
Gynecol Oncol. 54:254–258. 1994.
|
|
7
|
Takeshima N, Tabata T, Nishida H, et al:
Peripheral primitive neuroectodermal tumor of the vulva: report of
a case with imprint cytology. Acta Cytol. 45:1049–1052. 2001.
|
|
8
|
McCluggage WG, Sumathi VP, Nucci MR, et
al: Ewing family of tumors involving the vulva and vagina: report
of series of four cases. J Clin Pathol. 60:674–680. 2007.
|
|
9
|
Fong YE, López-Terrada D and Zhai QJ:
Primary Ewing sarcoma/peripheral primitive neuroectodermal tumor of
the vulva. Hum Pathol. 39:1535–1539. 2008.
|
|
10
|
Dehner LP: Primitive neuroectodermal tumor
and Ewing’s sarcoma. Am J Surg Pathol. 17:1–13. 1993.
|
|
11
|
de Alava E and Gerald WL: Molecular
biology of the Ewing’s sarcoma/primitive neuroectodermal tumor
family. J Clin Oncol. 18:204–213. 2000.
|
|
12
|
Dagher R, Pham TA, Sorbara L, et al:
Molecular confirmation of Ewing sarcoma. J Pediatr Hematol Oncol.
23:221–224. 2001.
|
|
13
|
Iwamoto Y: Diagnosis and treatment of
Ewing’s sarcoma. Jpn J Clin Oncol. 37:79–89. 2007.
|
|
14
|
Esiashvili N, Goodman M and Marcus RB Jr:
Changes in incidence and survival of Ewing sarcoma patients over
the past 3 decades: Surveillance Epidemiology and End Results data.
J Pediatr Hematol Oncol. 30:425–430. 2008.
|
|
15
|
Jain S and Kapoor G: Chemotherapy in
Ewing’s sarcoma. Indian J Orthop. 44:369–377. 2010.
|
|
16
|
Tanaka K, Matsunobu T, Sakamoto A, Matsuda
S and Iwamoto Y: High-dose chemotherapy and autologous peripheral
blood stem-cell transfusion after conventional chemotherapy for
patients with high-risk Ewing’s tumors. J Orthop Sci. 7:477–482.
2002.
|
|
17
|
Rodriguez-Galindo C, Billups CA, Kun LE,
et al: Survival after recurrence of Ewing tumors: the St. Jude
Children’s Research Hospital experience, 1979–1999. Cancer.
94:561–569. 2002.
|