Introduction
Multiple endocrine neoplasia type 1 (MEN1) is an
autosomal dominant cancer predisposition syndrome (1), caused by mutations in the MEN1
gene (2). The MEN1 gene is
located on chromosome 11q13 (2).
Previous studies of loss of heterozygosity (LOH) by microsatellite
analysis in tumor tissues of MEN1 patients have supported a tumor
suppressor function of the MEN1 gene (3–5).
Although patients with MEN1 syndrome are characterized by the
presence of tumors of the parathyroid gland, anterior pituitary and
endocrine pancreas (6), it has been
demonstrated that tumors may arise in over 20 different endocrine
and non-endocrine organs in these patients. Less common
manifestations in MEN1 patients include adrenocortical tumors,
foregut carcinoid tumors, such as thymic carcinoid, bronchial
carcinoid and gastric enterochromaffin-like tumors, and
cutaneous/mucosal or visceral abnormalities, such as facial
angiofibromas, lipomas, hypomelanotic macules, collagenomas and
meningiomas (6,7). There is also a frequent association
with thyroid tumors, however, this association should be considered
likely causal for the high incidence of thyroid abnormalities in
the general population (7).
The present study reports the case a patient with an
unusual combination of MEN1-associated tumors and breast cancer.
The patient exhibited major clinical manifestations of MEN1, such
as primary hyperparathyroidism, pituitary adenoma and pancreatic
endocrine carcinoma together with other tumors, including
adrenocortical adenoma, a thymic carcinoid tumor, papillary thyroid
carcinoma, uterine leiomyoma, lung hamatoma and breast cancer. Gene
analysis was performed for the MEN1, RET,
BRCA1 and BRCA2 genes to determine the association
between gene mutations and the development of tumors in the
patient. Written informed consent was obtained from the patient for
publication of this case report and accompanying images.
Case report
Patient
A 45-year-old female presented to the Daegu Catholic
University Hospital (Daegu, Korea) with a mass in the right breast
that had been present for the previous two months. The patient had
previously suffered no serious illnesses and had no known family
history of malignancy, including breast cancer. The patients’s
mother was known to have diabetes, but there was no known family
history of MEN1.
Physical examination and imaging
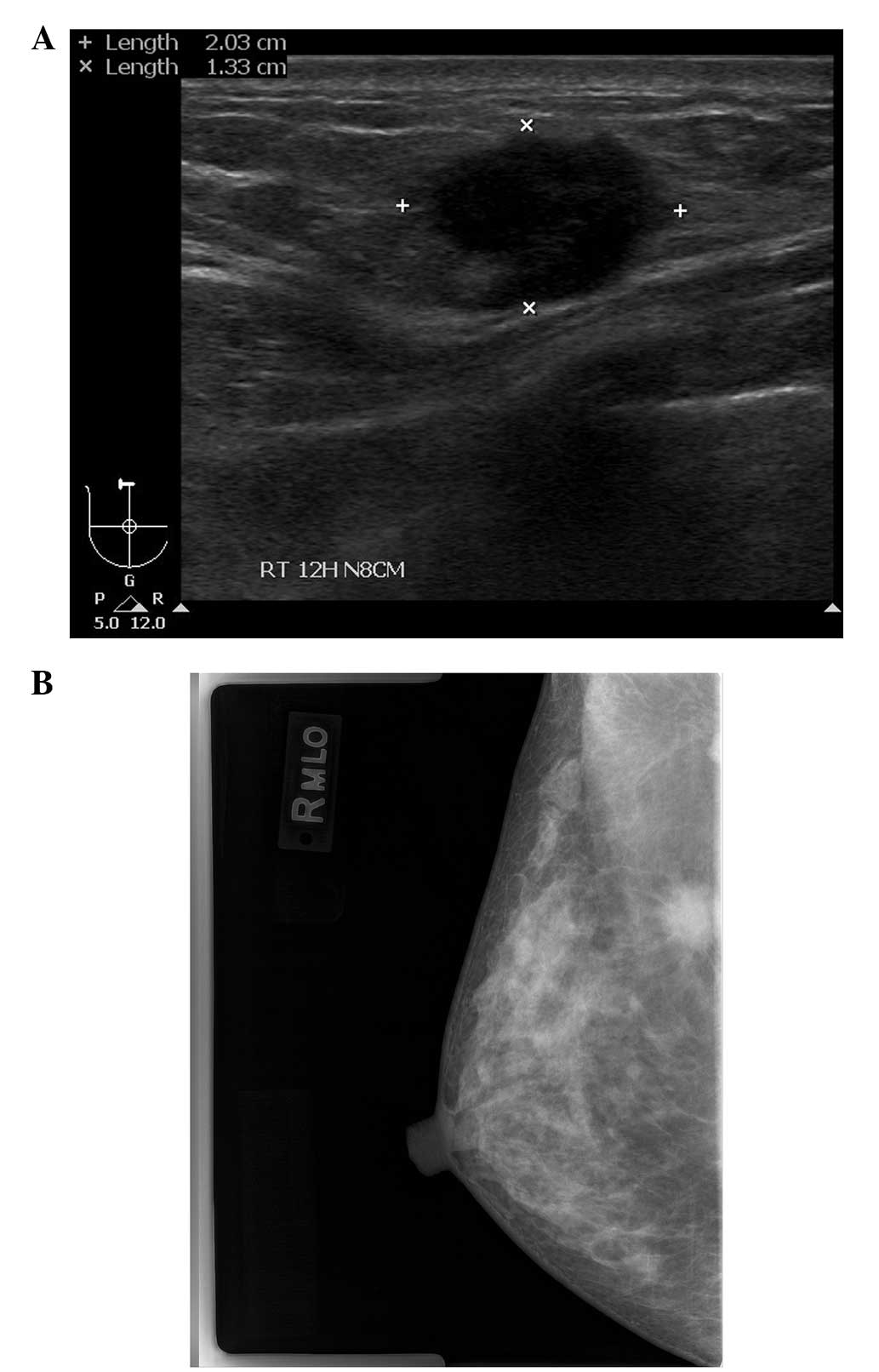
Upon physical examination, a fixed, firm mass, 2 cm
in diameter, was palpated without tenderness in the right breast.
There was no clinical evidence of regional lymphadenopathy.
Mammography revealed a spiculate hyperdense lesion in the upper
portion of the right breast (Fig.
1A). Ultrasonography (USG) revealed an irregularly-shaped
hypoechoic lesion in the right breast in accordance with the
finding of the mammography (Fig.
1B). The patient underwent an ultrasound-guided core needle
biopsy, which revealed the features of an invasive ductal
carcinoma. Radiological studies, including computed tomography (CT)
of the chest, magnetic resonance imaging (MRI) of the breast and
positron emission tomography-CT (PET-CT) of the torso were
conducted for pre-operative evaluation of the right breast cancer.
PET-CT showed metabolically active lesions in the right breast, the
anterior mediastinum, the peripancreatic area of the upper abdomen
and the left adrenal gland, which corresponded to the lesions
observed on the CT scan (Fig. 2).
In addition, the left thyroid gland and the endometrium of the
uterus showed mild FDG uptake on the PET-CT, and a suspicious
metastatic nodule of the lung was observed on the CT scan. The
findings of an additional abdominopelvic CT scan indicated a
neuroendocrine tumor of the pancreas, paraganglioma, a left adrenal
adenoma, gallstones and uterine subserosal myoma.
Laboratory results
Concomitantly, laboratory examinations revealed
hypercalcemia (11.8 mg/dl; normal range, 8.2–10.2 mg/dl),
hypophosphatemia (2.0 mg/dl; normal range, 2.5–4.5 mg/dl) and an
increased intact parathyroid hormone (iPTH) level of 340.8 pg/ml
(normal range, 12–72 pg/ml). The workup for the suspected MEN
syndrome revealed an increased basal plasma level of insulin-like
growth factor-1 (430 ng/ml; normal range, 124–290 ng/ml), prolactin
(43.9 ng/ml; normal range, 3–25 ng/ml) and calcitonin (286.3 pg/ml;
normal range, <10 pg/ml), and an increased 24-h urinary free
cortisol level (563.5 μg/24 h; normal range, 55.5–286.0). Basal
plasma levels of other hormones, including growth hormone, thyroid
stimulating hormone, adrenocorticotrophic hormone, gonadotrophic
hormone, cortisol, aldosterone, plasma rennin activity, gastrin,
insulin and urinary catecholamines, were all within normal
limits.
Imaging results
MRI brain scans showed a tumor of 1.4×0.9 cm in size
at the posterior aspect of the adenohypophysis, which was
indicative of a pituitary macroadenoma. USG of the neck revealed
relatively well-defined hypoechoic nodules in the bilateral thyroid
lobes. Fine-needle aspiration cytology for nodules at the inferior
pole of the bilateral thyroid lobes showed a few atypical
epithelial cells of suspected parathyroid origin.
Treatment and outcome
The patient underwent multiple pancreatic mass
enucleation, left adrenalectomy, cholecystectomy and hysterectomy.
The pathological diagnosis was of calcitonin-producing pancreatic
endocrine carcinoma for the pancreatic mass, adrenal cortical
adenoma for the adrenal mass, cholelithiasis and uterine leiomyoma
with adenomyosis, respectively. A month later, a right breast
lumpectomy with right axillary lymph node dissection, total
thyroidectomy, parathyroidectomy, extended thymectomy and wedge
resection of the lung were performed simultaneously. The
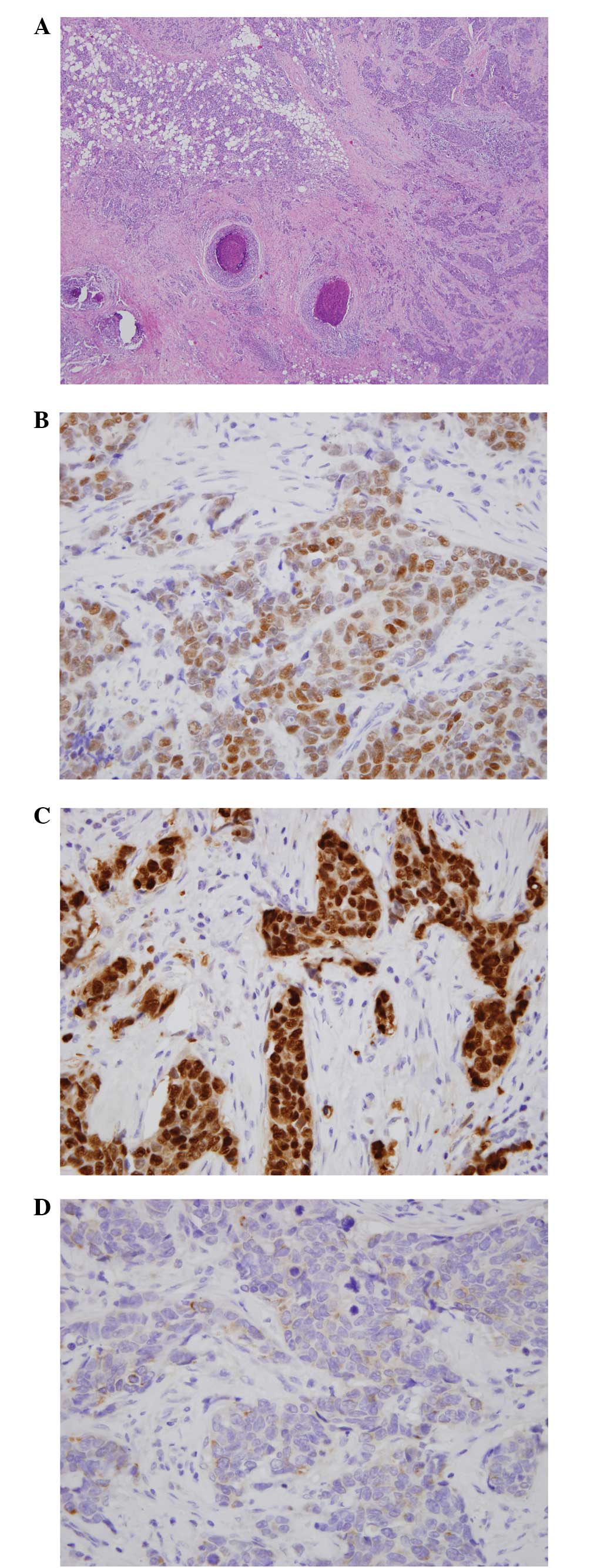
pathological diagnosis of the resected breast was of an invasive
ductal carcinoma associated with ductal carcinoma in situ
demonstrating estrogen receptor (ER)-positive, progesterone
receptor-positive and HER2/neu proliferation-negative breast
cancer, and metastatic carcinoma was detected in the right axillary
lymph nodes (Fig. 3). The
pathological diagnosis of nodules in the thyroid gland, parathyroid
gland, anterior mediastinal mass and lung nodule were bilateral
papillary thyroid carcinomas, not medullary carcinoma, and
parathyroid adenomas, a thymic carcinoid tumor and lung hamatoma,
respectively. Subsequent to the surgery, the serum calcium levels
and the iPTH decreased to within the normal range. The suspicious
pituitary adenoma remained untreated and has not since changed in
size in 2 years of follow-up examinations. Following the surgery,
the patient received adjuvant chemotherapy with 4 cycles of
Adriamycin and cyclophosphamide, followed by 4 cycles of docetaxel
and then radiation therapy to the right chest and axilla. The
patient is currently undergoing anti-estrogen therapy using
tamoxifen, and has exhibited no evidence of local tumor recurrence
or distant metastases in the 2 years since the surgery.
Mutational analysis
Given the clinical impression of combined MEN1 and
MEN2A based on the clinical manifestations of the patient,
confirmatory genetic testing for the MEN1, as well as the
RET gene was performed. Also, BRCA1 and BRCA2
genetic testing was performed to determine the association between
gene mutations and the development of other tumors, including
breast cancer.
Once informed consent had been obtained, peripheral
blood samples were collected from the patient. Genomic DNA was
extracted from blood using a commercial kit (Wizard Genomic DNA
Purification kit; Promega, Madison, WI, USA). Polymerase chain
reaction and mutational analyses of the genes were performed as
previously described (8–10). All coding exons for the MEN1
gene, and exons 10, 11, 13, 14, 15 and 16 of the RET
proto-oncogene were analyzed by direct sequencing. The 22 exons and
the exon-intron boundaries of the BRCA1 gene and the 26
exons and the exon-intron boundaries of the BRCA2 gene were
analyzed by direct sequencing. DNA sequencing was performed on the
pretreated PCR product using an automated direct sequence analyzer
(ABI PRISM 3100 Genetic Analyzer; Applied Biosystems, Foster City,
CA, USA).
Results
The MEN1 gene germline mutational analysis
revealed a 5-bp duplication in exon 3, namely, c.196_200dupAGCCC,
which resulted in a frameshift mutation of the MEN1 gene.
This mutation is one of the known germ-line mutations of the
MEN1 gene in MEN1 patients (11). In addition, a polymorphism of the
MEN1 gene was detected at codon 423 in exon 10 of the
MEN1 gene, with substitution of a cytidine to a thymidine
(C423T), which did not cause a change of amino acid. Mutation
analysis for the RET, BRCA1 and BRCA2 genes
showed a polymorphism of the RET and BRCA1 genes, but
no significant mutation was detected in this patient.
Discussion
The present study reports the case of a patient with
MEN1-associated tumors and breast cancer, in which we identified
germline mutations in MEN1, but not in BRCA1/2.
Although increasing evidence for MEN1-associated
non-endocrine tumors has been reported, there are limited data on
the association of breast cancer with MEN1. To the best of our
knowledge, there have been two reports of MEN1 associated with
breast cancer regardless of BRCA1/2 germline mutations
(12,13). Honda et al (12) reported a case with an unusual
combination of primary hyperparathyroidism, primary aldosteronism
and breast cancer, in a patient with a germline MEN1 gene
mutation, which is regarded as a benign polymorphism and loss of
heterozygosity (LOH) of the MEN1 locus in the DNA from
breast cancer tissue. The study hypothesized that the clinical
spectrum of MEN1 might include breast cancer. Recently, Inic et
al (13) also reported the case
of a patient with breast cancer and MEN1. Several other studies
have also described cases of patients with MEN1 and a family
history of breast cancer, however, in these studies, the breast
cancer was caused by mutations of the BRCA1/2 gene not the
MEN1 gene (14,15). Papi et al (14) reported the cases of carriers of both
the MEN1 and BRCA1 germline mutations, who had a
classical MEN1 phenotype with a family history of breast cancer.
Ghataorhe et al (15)
reported the case of a patient with both the MEN1 and
BRCA2 germline mutations, who had MEN1 and a family history
of male breast cancer.
The MEN1 gene responsible for MEN1 acts as a
tumor suppressor gene (16), and
tumors in MEN1 arise through the two-hit mechanism (3). The first hit is a germline mutation,
and the second hit is a somatic inactivation of the remaining
wild-type allele in a single cell of certain tissues, which
initiates neoplastic transformation (17). A wide variety of germline mutations
of the MEN1 gene have been identified to date (11,18).
These observed mutations are scattered throughout the entire coding
region and include nonsense, missense and frameshift mutations
(11). In the present study the
germline mutational analysis revealed a frameshift mutation in exon
3 of the MEN1 gene, which is a known mutation of the
MEN1 gene associated with MEN1 syndrome (11,18).
Several studies have indicated that mutation type or location
within MEN1 may be associated with clinical presentation
(19,20). However, there is no apparent
genotype-phenotype correlation (7,11).
Although 196_200dupAGCCC, the MEN1 germline mutation
detected in the present study, has previously been reported in
MEN1-related disorders (21–23),
the clinical manifestations of the patient in the present study are
different from those of previous studies, which indicates a lack of
genotype-phenotype correlation.
The product of the MEN1 gene, menin, is a
nuclear protein whose interaction with several nuclear proteins
indicates a role in transcriptional regulation (24–26).
Previous studies support a role for MEN1 in the control of
cell growth and differentiation, and in sensing or repairing DNA
damage (27–30). The loss of menin function in a tumor
precursor cell is involved in the mechanism for tumor formation in
MEN1 (1,20). In this regard, there are several
possible mechanisms of involvement for MEN1 in breast cancer
formation. Menin has been proposed to be involved in signaling
pathways that have a role in breast cancer formation, and it may
also control cell cycle progression and genomic integrity (1). Honda et al (12) hypothesized that an alteration of the
MEN1 gene with LOH and/or another tumor suppressor gene
located in the MEN1 locus on chromosome 11q13 may be
involved in the development of breast cancer without somatic gene
mutations. Data are conflicting as to how the loss of menin-ERα
interaction is associated with breast carcinogenesis. Menin can
directly interact with the ERα in a hormone-dependent manner
(31). Also, menin has a
demonstrable role as a coactivator for ERα-mediated transcription
by increasing the methylation of lysine 4 of histone 3 and the
consequent transcription of the trefoil factor-1 (TFF1) gene
(26,31). The product of TFF1 is
estrogen-induced breast cancer-associated peptide, and this is
indicated to be involved in breast carcinogenesis and a variety of
other tumor progression mechanisms (31–35).
Normal mammary tissue expresses little or no TFF1 protein
expression in normal breast ducts (36,37),
and TFF1 expression is increased and positively associated with
ER-positive tumors in breast cancer (35). Several studies have shown that the
protein expression of TFF1 is associated with an improved prognosis
and inversely associated with histological grade (33,35).
In summary, the current study presented the rare
case of a patient with MEN1 associated with breast cancer, in which
a germline mutation of the MEN1 gene was detected. In this
patient, MEN1 syndrome may have predisposed the patient to
developing breast cancer. However, there have been few studies
regarding the association between breast cancer and MEN1 syndrome,
and further studies and additional case reports are required to
clarify this connection.
References
|
1
|
Busygina V and Bale AE: Multiple endocrine
neoplasia type 1 (MEN1) as a cancer predisposition syndrome: clues
into the mechanisms of MEN1-related carcinogenesis. Yale J Biol
Med. 79:105–114. 2006.
|
|
2
|
Chandrasekharappa SC, Guru SC, Manickam P,
et al: Positional cloning of the gene for multiple endocrine
neoplasia-type 1. Science. 276:404–407. 1997.
|
|
3
|
Larsson C, Skogseid B, Oberg K, Nakamura Y
and Nordenskjöld M: Multiple endocrine neoplasia type 1 gene maps
to chromosome 11 and is lost in insulinoma. Nature. 332:85–87.
1988.
|
|
4
|
Thakker RV, Bouloux P, Wooding C, et al:
Association of parathyroid tumors in multiple endocrine neoplasia
type 1 with loss of alleles on chromosome 11. N Engl J Med.
321:218–224. 1989.
|
|
5
|
Farnebo F, Teh BT, Kytölä S, et al:
Alterations of the MEN1 gene in sporadic parathyroid tumors. J Clin
Endocrinol Metab. 83:2627–2630. 1998.
|
|
6
|
Brandi ML, Gagel RF, Angeli A, et al:
Guidelines for diagnosis and therapy of MEN type 1 and type 2. J
Clin Endocrinol Metab. 86:5658–5671. 2001.
|
|
7
|
Romei C, Pardi E, Cetani F and Elisei R:
Genetic and clinical features of multiple endocrine neoplasia types
1 and 2. J Oncol. 2012:7050362012.
|
|
8
|
Morelli A, Falchetti A, Martineti V, et
al: MEN1 gene mutation analysis in Italian patients with multiple
endocrine neoplasia type 1. Eur J Endocrinol. 142:131–137.
2000.
|
|
9
|
Chung YJ, Kim HH, Kim HJ, et al: RET
proto-oncogene mutations are restricted to codon 634 and 618 in
Korean families with multiple endocrine neoplasia 2A. Thyroid.
14:813–818. 2004.
|
|
10
|
Kim BY, Lee DG, Lee KR, et al:
Identification of BRCA1 and BRCA2 mutations from Korean breast
cancer patients using denaturing HPLC. Biochem Biophys Res Commun.
349:604–610. 2006.
|
|
11
|
Giraud S, Zhang CX, Serova-Sinilnikova O,
et al: Germ-line mutation analysis in patients with multiple
endocrine neoplasia type 1 and related disorders. Am J Hum Genet.
63:455–467. 1998.
|
|
12
|
Honda M, Tsukada T, Horiuchi T, et al:
Primary hyperparathyroidism associated with aldosterone-producing
adrenocortical adenoma and breast cancer: Relation to MEN1 gene.
Intern Med. 43:310–314. 2004.
|
|
13
|
Inic ZM, Inic M, Dodic R, Pupic G and
Damjanovic S: Breast cancer in a patient with multiple endocrine
neoplasia type 1 (MEN1): A case report and review of the
literature. J Clin Oncol. 30(suppl): abstract 221136. 2012.
|
|
14
|
Papi L, Palli D, Masi L, et al: Germline
mutations in MEN1 and BRCA1 genes in a woman with familial multiple
endocrine neoplasia type 1 and inherited breast-ovarian cancer
syndromes: a case report. Cancer Genet Cytogenet. 195:75–79.
2009.
|
|
15
|
Ghataorhe P, Kurian AW, Pickart A, et al:
A carrier or both MEN1 and BRCA2 mutations: case report and review
of the literature. Cancer Genet Cytogenet. 179:89–92. 2007.
|
|
16
|
Chandrasekharappa SC and Teh BT:
Functional studies of the MEN1 gene. Functional studies of the MEN1
gene. J Intern Med. 253:606–615. 2003.
|
|
17
|
Knudson AG Jr: Mutation and cancer:
statistical study of retinoblastoma. Proc Natl Acad Sci USA.
68:820–823. 1971.
|
|
18
|
Lemos MC and Thakker RV: Multiple
endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations
reported in the first decade following identification of the gene.
Hum Mutat. 29:22–32. 2008.
|
|
19
|
Kouvaraki MA, Lee JE, Shapiro SE, et al:
Genotype-phenotype analysis in multiple endocrine neoplasia type 1.
Arch Surg. 137:641–647. 2002.
|
|
20
|
Lips CJ, Dreijerink KM and Höppener JW:
Variable clinical expression in patients with a germline MEN1
disease gene mutation: clues to a genotype-phenotype correlation.
Clinics (Sao Paolo). 67(Suppl 1): 49–56. 2012.
|
|
21
|
Ellard S, Hattersley AT, Brewer CM and
Vaidya B: Detection of an MEN1 gene mutation depends on clinical
features and supports current referral criteria for diagnostic
molecular genetic testing. Clin Endocrinol (Oxf). 62:169–175.
2005.
|
|
22
|
Klein RD, Salih S, Bessoni J and Bale AE:
Clinical testing for multiple endocrine neoplasia type 1 in a DNA
diagnostic laboratory. Genet Med. 7:131–138. 2005.
|
|
23
|
Park JH, Kim IJ, Kang HC, et al: Germline
mutations of the MEN1 gene in Korean families with multiple
endocrine neoplasia type 1 (MEN1) or MEN1-related disorders. Clin
Genet. 64:48–53. 2003.
|
|
24
|
Agarwal SK, Guru SC, Heppner C, et al:
Menin interacts with the AP1 transcription factor JunD and
represses JunD-activated transcription. Cell. 96:143–152. 1999.
|
|
25
|
Heppner C, Bilimoria KY, Agarwal SK, et
al: The tumor suppressor protein menin interacts with NF-kappaB
proteins and inhibits NF-kappaB-mediated transactivation. Oncogene.
20:4917–4925. 2001.
|
|
26
|
Kim H, Lee JE, Cho EJ, Liu JO and Youn HD:
Menin, a tumor suppressor, represses JunD-mediated transcriptional
activity by association with an mSin3A-histone deacetylase complex.
Cancer Res. 63:6135–6139. 2003.
|
|
27
|
Itakura Y, Sakurai A, Katai M, et al:
Enhanced sensitivity to alkylating agent in lymphocytes from
patients with multiple endocrine neoplasia type 1. Biomed
Pharmacother. 54(Suppl 1): 187s–190s. 2000.
|
|
28
|
Jin S, Mao H, Schnepp RW, et al: Menin
associates with FANCD2, a protein involved in repair of DNA damage.
Cancer Res. 63:4204–4010. 2003.
|
|
29
|
Binz SK, Sheehan AM and Wold MS:
Replication protein A phosphorylation and the cellular response to
DNA damage. DNA Repair (Amst). 3:1015–1024. 2004.
|
|
30
|
Kim YS, Burns AL, Goldsmith PK, et al:
Stable overexpression of MEN1 suppresses tumorigenicity of RAS.
Oncogene. 18:5936–5942. 1999.
|
|
31
|
Dreijerink KM, Mulder KW, Winkler GS,
Höppener JW, Lips CJ and Timmers HT: Menin links estrogen receptor
activation to histone H3K4 trimethylation. Cancer Res.
66:4929–4935. 2006.
|
|
32
|
Prest SJ, May FE and Westley BR: The
estrogen-regulated protein, TFF1, stimulates migration of human
breast cancer cells. FASEB J. 16:592–594. 2002.
|
|
33
|
Corte MD, Tamargo F, Alvarez A, et al:
Cytosolic levels of TFF1/pS2 in breast cancer: Their relationship
to clinical-pathological parameters and their prognostic
significance. Breast Cancer Res Treat. 96:63–72. 2006.
|
|
34
|
Bauche E, Etique N, Alpy F, et al:
Deficiency in trefoil factor 1 (TFF1) increases tumorigenicity of
human breast cancer cells and mammary tumor development in
TFF1-knockout mice. Oncogene. 30:3261–3273. 2011.
|
|
35
|
Amiry N, Kong X, Muniraj N, et al: Trefoil
factor-1 (TFF1) enhances oncogenicity of mammary carcinoma cells.
Endocrinology. 150:4473–4483. 2009.
|
|
36
|
Hähnel E, Robbins P and Hähnel R:
Expression of the pS2 gene in normal breast tissue. Breast Cancer
Res Treat. 28:295–297. 1993.
|
|
37
|
Poulsom R, Hanby AM, Lalani EN, Hauser F,
Hoffmann W and Stamp GW: Intestinal trefoil factor (TFF 3) and pS2
(TFF 1), but not spasmolytic polypeptide (TFF 2) mRNAs are
co-expressed in normal, hyperplastic, and neoplastic human breast
epithelium. J Pathol. 183:30–38. 1997.
|