Introduction
Metformin is one of the first-line drugs used for
type 2 diabetes treatment and has been used for over half a
century. Recent studies found that metformin not only effectively
reduced hepatic glucose production and increased insulin
sensitivity, but that it also was effective in decreasing the risk
of cancer in patients with type 2 diabetes, inhibiting the growth
of cancer cells and enhancing the effects of chemotherapeutic drugs
(1).
The potentially beneficial effects of metformin
against cancer are believed to be mediated mainly by 5′-adenosine
monophosphate-activated protein kinase (AMPK), a well-conserved
energy sensor that plays a key role in the regulation of protein
and lipid metabolism in response to changes in fuel availability.
Activated AMPK inhibits cell growth and proliferation, and
therefore antagonizes cancer cell growth (2).
However, more recent data have indicated that
metformin can inhibit proliferation and sensitize cancer cells to
anticancer drugs through the inhibition of HO-1, by targeting
Raf-ERK-Nrf2 signaling in an AMPK-independent manner. Therefore,
the mechanisms of the suppression of cancer cell growth by
metformin remain unclear (3).
Previous studies have demonstrated the inhibition of
metformin on cancer cells (3–7). In
the present study, we aimed to understand the anti-tumor molecular
mechanisms of metformin.
Materials and methods
Cell line and culture conditions
The human mammary carcinoma T47D cell line was
purchased from American Type Culture Collection (Manassas, VA,
USA). The T47D cells were maintained with RPMI 1640 (Gibco,
Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with
10% fetal calf serum (Hyclone, Thermo Scientific, Waltham, MA,
USA), penicillin (100 U/ml) and streptomycin (100 μg/ml). The cells
were cultured at 37°C in 5% CO2. Additionally, 0.25%
trypsin was purchased from Gibco. Metformin was purchased from
Sigma-Aldrich (St. Louis, MO, USA).
Flow cytometry analysis
The T47D cells were cultured in 6-well plates and
treated with 4 mM metformin for 48 h, then fixed by 70% ethyl
alcohol, which was subsequently removed by centrifugation at 250 ×
g for 5 min. RNase A was added in for 30 min. Following propidium
iodide staining, the cell cycle was detected with a FACSCalibur
flow cytometer (BD Biosciences, San Jose, CA, USA).
Western blotting analysis
Western blotting was conducted, as previously
described (7,8). For histone extraction, the cells were
lysed with ice-cold NETN buffer containing 10 mM NaF and 50 mM
β-glycerophosphate, and following centrifugation at 250 × g for 5
min, the remaining pellets were washed twice with ice-cold PBS and
then treated with 200 μl 0.2 HCl. The supernatants were neutralized
with 40 μl 1N NaOH, and the sample was loaded onto 12.5% SDS-PAGE
gels for western blotting with the indicated antibodies. Monoclonal
rabbit anti-human anti-phospho-acetyl-CoA carboxylase (Ser79)
(pACC1), monoclonal rabbit anti-human anti-phospho-AMPKα
(Thr172)(p-AMPKα1), monoclonal rabbit anti-human
anti-ubiquityl-histone H2B (Lys120) (H2B K120ub) and rabbit
monoclonal anti-human anti-AMPKα1 were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA), with the exception
of monoclonal mouse anti-human β-actin, was purchased from
Sigma-Aldrich.
Quantitative (q)PCR
Total mRNA was isolated from the cells with TRIzol
(Invitrogen Life Technolgies), and then complementary DNA (cDNA)
was synthesized from 500 ng total RNA with the
PrimeScript® 1st Strand cDNA Synthesis kit (Takara
Biotechnology, Dalian, China). qPCR was performed on a 7500RT-PCR
System (Applied Biosystems, Foster City, CA, USA) using the SYBR
Green detection system with the following program: 95°C for 5 min
for 1 cycle, followed by 95°C for 30 sec and 60°C for 45 sec for 40
cycles. The relative expression of the target genes, including p21,
cyclin D1, Tulp4 and β-actin, was represented by 2−ΔΔCT.
All samples were normalized to the β-actin mRNA levels, and the
relative expression of the mRNA of every treatment group was
calculated. The experiment was duplicated three times. The primer
sequences that were used are as follows: Cyclin D1 forward,
5′-ACGCTTCCTCTCCAGAGTGAT-3′ and reverse, 5′-TTGACTCCAGCAGGGCTT-3′;
Tulp4 forward, 5′-GGGCCACAATAGCGAGGTT-3′ and reverse,
5′-CCACACGAATATGCCTCCGT-3′; p21 forward, 5′-TGTCCGTCAGAACCCATGC-3′
and reverse, 5′-AAAGTCGAAGTTCCATCGCTC-3′; and β-actin forward,
5′-GTCTGCCTTGGTAGTGGATAATG-3′ and reverse,
5′-TCGAGGACGCCCTATCATGG-3′.
Cell proliferation
The CellTiter-Blue assay kit (Promega, Southampton,
UK) was used to measure the number of cells, according to the
manufacturer’s instructions. Briefly, in a 96-well plate, the cells
were washed three times with PBS and 20 μl of CellTiter-Blue
reagent (Promega) was added. The plate was incubated for 4 h
protected from light, and the fluorescence intensity was recorded
(excitation, 560 nm; emission, 590 nm) on a Tecan M200 microplate
reader (Tecan Australia, Port Melbourne, Vic, Australia).
Small interfering (si)RNA
transfections
siRNA targeting AMPKα1 and a siRNA transfection
reagent were obtained from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). The T47D cells, grown to 50% confluence, were
transfected with AMPKα1siRNA or a non-specific control siRNA. After
48 h, the cells were used for experimentation.
Statistical analysis
The results are presented as the mean ± standard
deviation. The statistical analysis was performed with SPSS 13.0
software (SPSS, Inc., Chicago, IL, USA). For comparisons between
multiple groups, a one-way analysis of variance was used, while for
a comparison between two groups, the SNK method was used. For
comparisons between the treatment and control groups, Dunnett’s
t-test was used, and for the analysis of differences between
groups, the Student’s t-test was used. P<0.05 was considered to
indicate a statistically significant difference.
Results
Metformin inhibits breast cancer cell
proliferation and induces cell cycle arrest
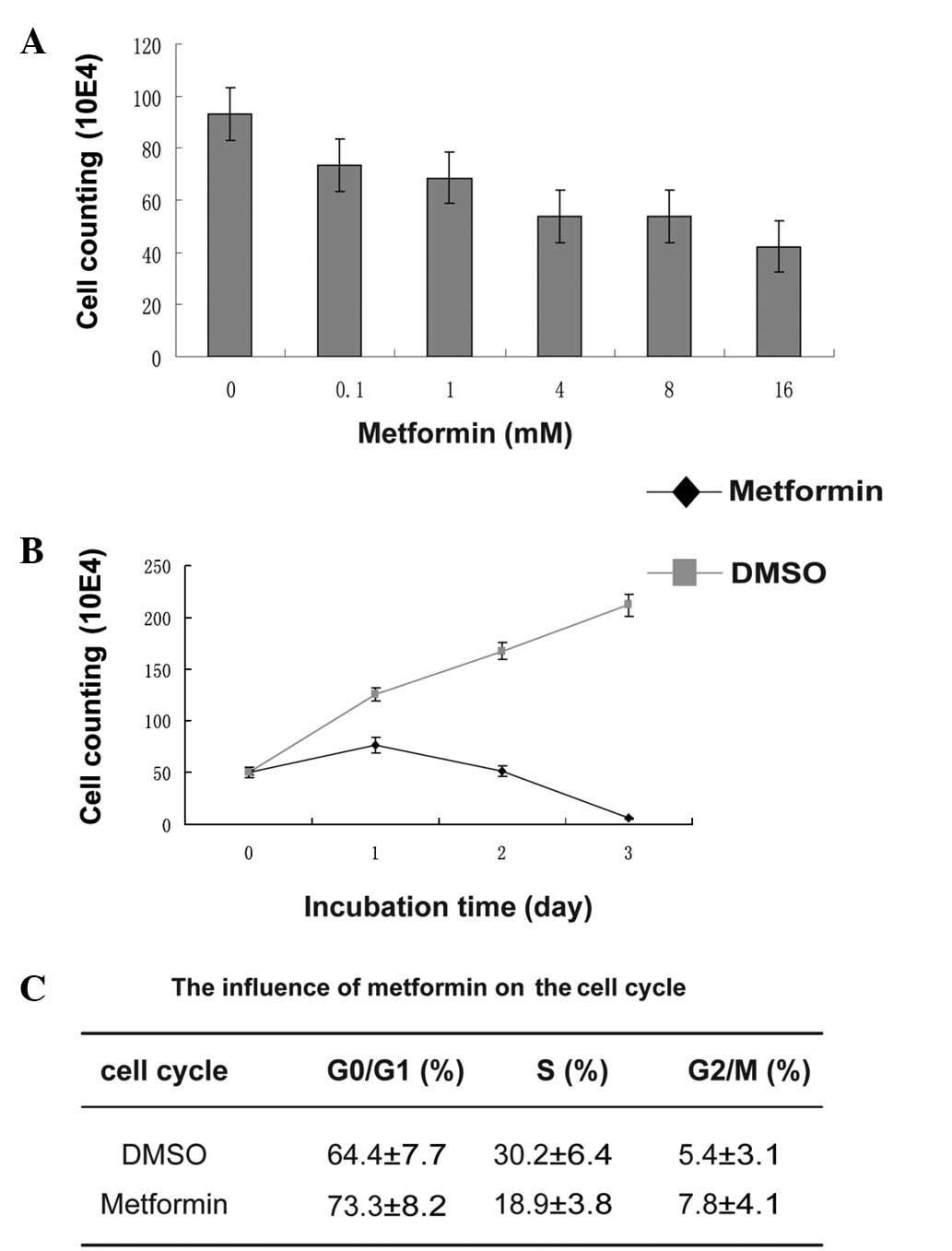
To detect the effect of metformin on breast cancer
cell proliferation, the T47D cells were treated with increasing
doses of metformin for 24 h. The inhibition of T47D cell growth by
metformin occurred in a dose-dependent manner (Fig. 1A). The cells were almost completely
killed by 8 mM metformin for 72 h (Fig.
1B). The cell cycle of the T47D cells was analyzed by flow
cytometer following treatment with 4 mM metformin for 48 h; the
ratio of the cells in G0/G1 phase increased
from 64.4 to 73.3%, while that of cells in the S-phase dropped from
30.2 to 18.9% (Fig. 1C). These
results showed that metformin significantly inhibited the
proliferation of the cells and induced cell cycle arrest.
Metformin activates AMPK and inhibits
histone H2B monoubiquitination and downstream gene
transcription
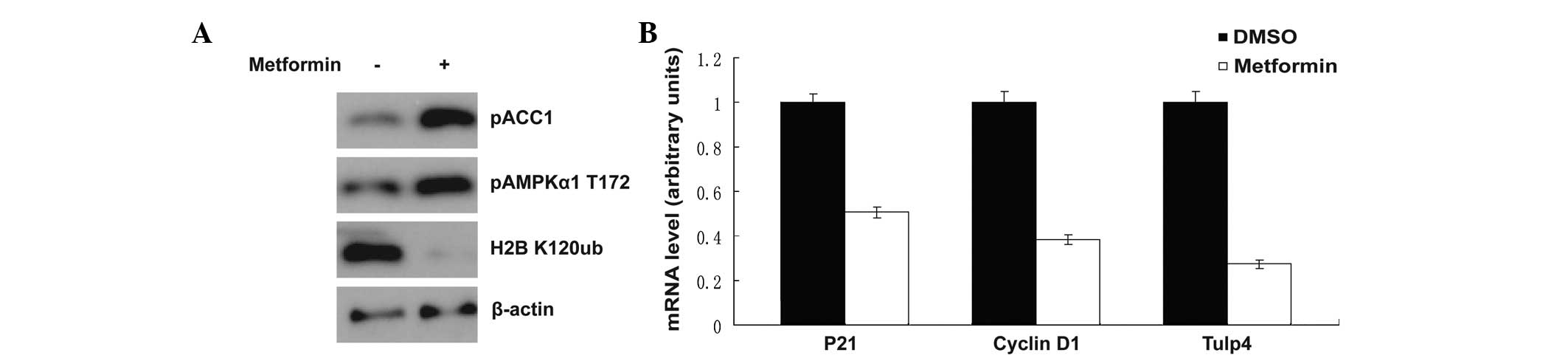
Metformin is a well-known AMPK activator. Upon
treatment with metformin, AMPK was activated, as shown in Fig. 2A, AMPK threonine 172 phosphorylation
was increased and the phosphorylation of acetyl-CoA carboxylase at
serine 79 was markedly enhanced (Fig.
2A). This result was consistent with the results of previous
studies (5–9). Notably, histone H2B monoubiquitination
at lysine 120 was inhibited at the same time. It was reported
previously that when the cells were suffering a shortage of
glucose, the H2B monoubiquitination at lysine 120 was also
inhibited (10). The H2B
monoubiquitination at lysine 120 was associated with the
transcription of multiple downstream target genes (11,12).
In the present study, transcription of the downstream genes,
including p21, cyclin D1 and Tulp4, was detected by qPCR, and the
mRNA level was shown to be decreased significantly following
exposure to metformin (Fig.
2B).
Metformin inhibits breast cancer cell
proliferation, which is dependent on AMPK
Since metformin efficiently activated the AMPK
signal transduction pathway, the study also detected whether the
inhibition of cell proliferation by metformin was AMPK-dependent.
AMPKα1 protein was specifically knocked down by AMPKα1 siRNA
(Fig. 3A). The AMPKα1 siRNA and
control groups were then treated with 4 mM metformin for 24 h. The
inhibition of T47D cell proliferation by metformin was found to be
less effective in the AMPKα1 siRNA group (Fig. 3B). This experiment confirmed that
the inhibition of cell proliferation by metformin was
AMPK-dependent.
Discussion
An increasing number of studies are showing that
diabetic patients treated with metformin have a lower incidence of
cancer compared with those on other treatments (6,13,14).
Another large case-control study has indicated that metformin may
somewhat reduce the incidence of pancreatic cancer (15). Early-stage clinical trials are
currently underway to investigate the potential of metformin to
prevent an array of cancers, including colorectal, prostate,
endometrial and breast cancer (16–18).
However, the underlying mechanism remains to be fully
elucidated.
A few of the beneficial effects of metformin have
been shown to work through the activation of AMPK. Treatment with
metformin results in the activation of AMPK in in vitro and
in vivo experiments, and the activation of AMPK is well
known to inhibit the expression of gluconeogenic genes and to
promote the expression of enzymes required for fatty acid oxidation
(19–21).
However, it has also been reported that in
AMPK-knockout cells, metformin works through inhibition of HO-1 by
targeting Raf-ERK-Nrf2 signaling, which indicates that a novel
mechanism is present (3).
The results of the present study indicated that
metformin significantly inhibited the proliferation of the breast
cancer cells and induced cell cycle arrest in an AMPK-dependent
manner. This is consistent with the results of previous studies.
Further molecular mechanism studies showed that metformin activates
the AMPK signal transduction pathway, promoting the phosphorylation
of ACC1, so that the synthesis of fatty acids of carcinoma cells is
inhibited. As a result, the proliferation of the cells was reduced.
Unexpectedly, metformin was able to inhibit histone H2B K120-ub,
which is related to the transcription of downstream target genes,
such as p21 and cyclin D1, which function as regulators of the cell
cycle (11,12). The results of qPCR detection
indicated that the transcription of p21, Tulp4 and cyclin D1 was
inhibited by metformin. This partially explained the mechanism by
which metformin blocks the cell cycle.
The present study revealed the possible novel
anticancer mechanism of metformin. However, the manner by which
metformin inhibits H2B monoubiquitination remains unknown. It is
presumed that metformin can activate AMPK, which phosphorylates a
certain substrate. This substrate is capable of inhibiting the
histone ubiquitination mediated by E3 ubiquitin-protein ligase.
Further studies should be performed on the relevant molecular
mechanism involved.
Acknowledgements
This study was supported by the Natural Science
Foundation of Hubei province, China (grant no. 2008CDB168).
References
|
1
|
Li Q, Guo D, Dong Z, et al: Ondansetron
can enhance cisplatin-induced nephrotoxicity via inhibition of
multiple toxin and extrusion proteins (MATEs). Toxicol Appl
Pharmacol. 273:100–109. 2013.
|
|
2
|
Hardie DG: AMP-activated/SNF1 protein
kinases: conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–785. 2007.
|
|
3
|
Do MT, Kim HG, Khanal T, et al: Metformin
inhibits heme oxygenase-1 expression in cancer cells through
inactivation of Raf-ERK-Nrf2 signaling and AMPK-independent
pathways. Toxicol Appl Pharmacol. 271:229–238. 2013.
|
|
4
|
Lettieri Barbato D, Vegliante R, Desideri
E and Ciriolo MR: Managing lipid metabolism in proliferating cells:
New perspective for metformin usage in cancer therapy. Biochim
Biophys Acta. 1845:317–324. 2014.
|
|
5
|
Leverve XM, Guigas B, Detaille D, et al:
Mitochondrial metabolism and type-2 diabetes: a specific target of
metformin. Diabetes Metab. 29:6S88–6S94. 2003.
|
|
6
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005.
|
|
7
|
Anastasiou D: Metformin: a case of divide
and conquer. Breast Cancer Res. 15:3062013.
|
|
8
|
Isakovic A, Harhaji L, Stevanovic D, et
al: Dual antiglioma action of metformin: cell cycle arrest and
mitochondria-dependent apoptosis. Cell Mol Life Sci. 64:1290–1302.
2007.
|
|
9
|
Kemp BE, Stapleton D, Campbell DJ, et al:
AMP-activated protein kinase, super metabolic regulator. Biochem
Soc Trans. 31:162–168. 2003.
|
|
10
|
Sanders MJ, Grondin PO, Hegarty BD,
Snowden MA and Carling D: Investigating the mechanism for AMP
activation of the AMP-activated protein kinase cascade. Biochem J.
403:139–148. 2007.
|
|
11
|
Zhu B, Zheng Y, Pham AD, et al:
Monoubiquitination of human histone H2B: the factors involved and
their roles in HOX gene regulation. Mol Cell. 20:601–611. 2005.
|
|
12
|
Fujiki R, Hashiba W, Sekine H, et al:
GlcNAcylation of histone H2B facilitates its monoubiquitination.
Nature. 480:557–560. 2011.
|
|
13
|
Decensi A, Puntoni M, Goodwin P, et al:
Metformin and cancer risk in diabetic patients: a systematic review
and meta-analysis. Cancer Prev Res (Phila). 3:1451–1461. 2010.
|
|
14
|
Noto H, Goto A, Tsujimoto T and Noda M:
Cancer risk in diabetic patients treated with metformin: a
systematic review and meta-analysis. PLoS One. 7:e334112012.
|
|
15
|
Lee MS, Hsu CC, Wahlqvist ML, Tsai HN,
Chang YH and Huang YC: Type 2 diabetes increases and metformin
reduces total, colorectal, liver and pancreatic cancer incidences
in Taiwanese: a representative population prospective cohort study
of 800,000 individuals. BMC Cancer. 11:202011.
|
|
16
|
Belfiore A and Frasca F: IGF and insulin
receptor signaling in breast cancer. J Mammary Gland Biol
Neoplasia. 13:381–406. 2008.
|
|
17
|
Slomiany MG, Black LA, Kibbey MM, Tingler
MA, Day TA and Rosenzweig SA: Insulin-like growth factor-1 receptor
and ligand targeting in head and neck squamous cell carcinoma.
Cancer Lett. 248:269–279. 2007.
|
|
18
|
Weiss JM, Huang WY, Rinaldi S, et al:
IGF-1 and IGFBP-3: Risk of prostate cancer among men in the
Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial. Int
J Cancer. 121:2267–2273. 2007.
|
|
19
|
Dowling RJ, Zakikhani M, Fantus IG, Pollak
M and Sonenberg N: Metformin inhibits mammalian target of
rapamycin-dependent translation initiation in breast cancer cells.
Cancer Res. 67:10804–10812. 2007.
|
|
20
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006.
|
|
21
|
Shaw RJ, Bardeesy N, Manning BD, et al:
The LKB1 tumor suppressor negatively regulates mTOR signaling.
Cancer Cell. 6:91–99. 2004.
|