Introduction
Pituitary adenoma is the most common type of tumor
found in the sellar region. It is believed to account for 10–15% of
all primary brain tumors (1).
Gangliocytomas [World Health Organization (WHO) grade I) are rare
benign tumors of the sympathetic nerve fibers arising from neural
crest cells, which can grow wherever sympathetic nervous tissue is
found (1). Gangliocytoma commonly
occurs in adolescents and young adults (40–60%), but individuals of
all ages may be affected. Development of this tumor occurs at the
extracranial and intracranial sites, with an incidence rate of
~0.5% at the intracranial site (2).
The current study presents two cases of a hormone-free pituitary
adenoma with gangliocytoma in the sellar region.
Case reports
Case one
A 47-year-old female presented to the Huashan
Hospital (Fudan University, Shanghai, China) with blurred vision
that had been present for the past two years. The patient had no
obvious symptoms of headaches, nausea or vomiting, no notable
polydipsia or diuresis and no phantom pain, limb activity disorder
or body convulsions. However, two years prior to admittance, the
patient was diagnosed with amenorrhea. One year prior to
admittance, magnetic resonance imaging (MRI) revealed a lesion in
the sellar region.
Upon neurological examination, a diagnosis of
acromegaly was formed. The patient was able to fix and follow
objects with each eye. Light perception was only present in the
right eye, and the vision in the left eye was 0.2 decimal units.
The fundus was unremarkable and the pupils were regular and
reactive.
A computed tomography (CT) scan (Fig. 1) revealed a parenchymatous mass in
the sellar region, damage of the sclerotin of the anterior clinoid
process, partial obliterations of the suprasellar cistern and a
compressed anterior third ventricle and precornu. MRI (Fig. 2) showed a sellar lesion, with
heterogeneous enhancement on the contrast MRI.
A perimetry examination showed complete defects of
the right eye in the 30 degree field, and defects of the left eye
on the side of the temporal bone.
Pre-operative laboratory examinations found 1.21
mmol/l triiodothyrine (T3), 3.31 pmol/l free T3 (FT3), 10.28 pmol/l
free thyroxine (FT4), 64.7 pg/ml adrenocorticotropic hormone (ACTH)
and 8.9 mU/l growth hormone (GH). Post-operative laboratory
examinations found <0.84 mmol/l TT3, 2.42pmol/l FT3, 9.36 pmol/l
FT4, 19.0 pg/ml ACTH and 28.7 mU/l GH.
The patient was hospitalized due to a
space-occupying lesion found in the sellar region in January 2012,
and underwent sellar region tumor resection via the
trans-naso-sphenoid approach. During surgery, it was found that the
tumor exhibited a gray-red color and a heterogeneous texture.
Softly-textured tumor sections were excised and examined under
microscope, together with firmly-textured tumor sections, which
were hard to excise.
Post-operative pathological examination confirmed
the diagnosis of a hormone-free pituitary adenoma with
gangliocytoma [World Health Organization (WHO) grade I]. Under a
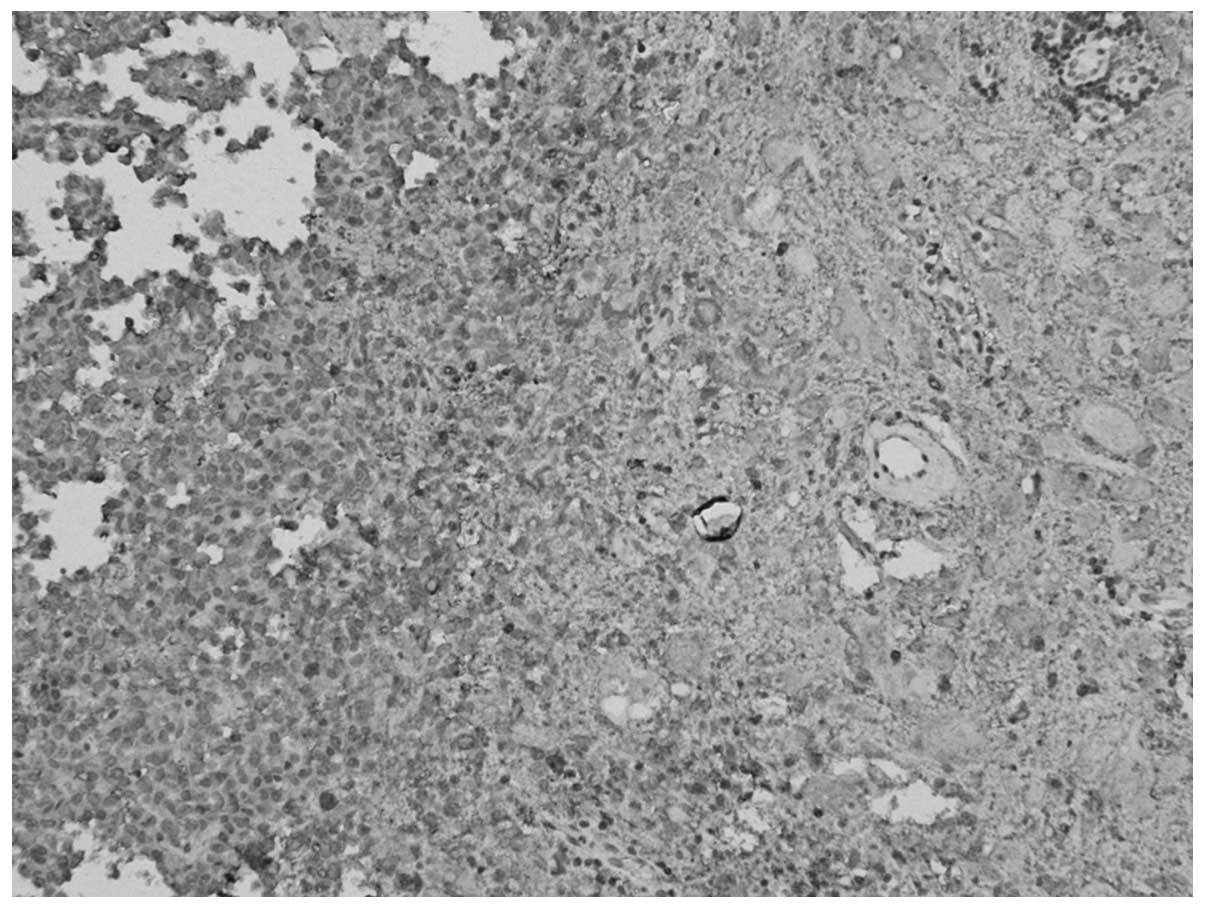
spectroscope, (Fig. 3) two types of
tumor components were found; certain areas showed a nest-shaped
distribution of the cell debris of the pituitary adenoma, and were
positively stained for Syn and negatively stained for GH,
luteinizing hormone (LH), prolactin (PRL), thyroid-stimulating
hormone (TSH), follicle-stimulating hormone (FSH) and ACTH. Other
areas showed a laminar distribution of ganglion cell-like neurons
and were positively stained for Syn and negatively stained for
cluster of differentiation 34. MIB-1 expression was <1%.
Subsequent to a three-month follow-up period, the
patient’s vision in the left eye remained at 0.2, while the vision
in the right eye increased to 0.1.
Case two
A 47-year-old female presented to the Huashan
Hospital with a lesion in the sellar region, which was found one
month previously on MRI examination due to a head trauma. The
patient typically had good vision and in recent days had no notable
polydipsia or diuresis and no phantom pain, limb activity disorder
or body convulsions. However, one year prior to admittance, the
patient was diagnosed with amenorrhea.
Upon neurological examination, the patient was able
to fix and follow objects with each eye, and the vision of each eye
was 0.8 decimal units. The fundus was unremarkable and the pupils
were regular and reactive.
A CT scan (Fig. 4)
showed that there was a space-occupying lesion in the sellar area,
which was situated on the right-hand side. The sellar area was
expanded and the end of the saddle had sunk. MRI (Fig. 5) revealed an irregular abnormal
signal measuring ~3.7×2.1 cm in size and marked heterogeneous
enhancement on the contrast MRI, which affected the right cavernous
sinus and surrounded the right internal carotid artery. The
pituitary stalk was compressed to the left, which caused
obliterations of the suprasellar cistern.
Perimetry and endocrinological examinations of the
eyes were normal.
The patient was hospitalized due to the presence of
a space-occupying lesion of the sellar region in March 2012, and
underwent a sellar region tumor resection via the
trans-naso-sphenoid approach. During surgery, it was found that the
tumor exhibited a gray-red color, a firm texture and an adequate
blood supply. The majority of the tumor was excised under a
microscope. Post-operative pathological examination confirmed the
diagnosis of a hormone-free pituitary adenoma with gangliocytoma
(WHO grade I).
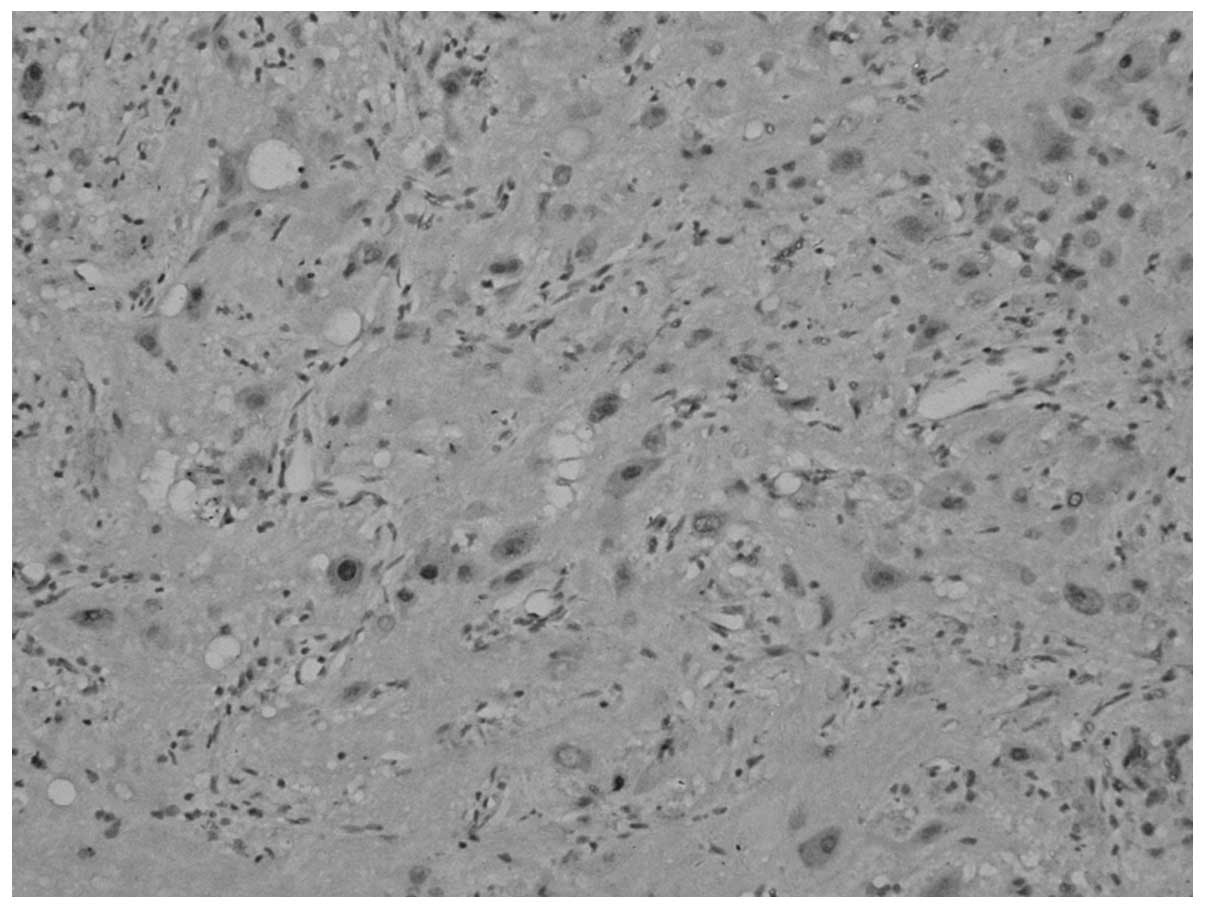
Under a spectroscope (Fig. 6), two types of tumor components were
found; certain areas of the tumor displayed a nest-shaped
distribution of the cell debris of the pituitary adenoma, and other
areas showed a laminar distribution of ganglion cell-like neurons
with collagenous fibrosis and mild calcification.
Immunohistochemical analysis of the cells of the pituitary adenoma
revealed the following: Positive staining for Syn, and negative
staining for GH, LH, PRL, TSH, FSH and ACTH. MIB-1 expression was
<1%. The ganglion cell-like neurons were positive for Syn and
NeuN, and MIB-1 expression was <1%.
Subsequent to a one-month follow-up period, the
patient’s vision was restored to the same quality as prior to the
surgery, with no significant changes observed. The patient was in a
good condition and experienced no specific discomfort.
Discussion
Pituitary adenoma is a common benign tumor of the
sellar region, which typically originates from the anterior
pituitary, while gangliocytoma is a rare benign tumor of
neuroblastic origin, which originates from the posterior pituitary
(1). However, these mixed pituitary
tumors have been rarely reported worldwide.
Gangliocytoma (WHO grade I) originates from the
neuron, and is attributable to neuronal and mixed neuronal-glial
tumors. Neuroblastic tumors can be broadly subcategorized as
neuroblastoma, ganglioneuroblastoma or gangliocytoma (2). The three tumors differ in their degree
of cellular and extracellular maturation. The most benign tumor is
the gangliocytoma, which is composed entirely of neural elements,
including mature ganglion cells and schwannian stroma, and does not
contain neuroblasts, intermediate cells or mitotic figures
(3).
Gangliocytoma commonly occurs in adolescents and
young adults (40–60%), but individuals of all ages can be affected.
Development of this tumor is common at a young age and occurs at
the extracranial and intracranial sites, with an incidence rate of
~0.5% at the intracranial site (3).
Gangliocytoma in the intracranial site is prone to be distributed
in the end of the third ventricle and in the frontal and temporal
lobes (4), and is rarely found in
the sellar region. Greenfield (5)
reported one type of tumor that originated from the anterior and
posterior pituitary of the sellar region in 1919, which was
originally named a choristoma. Towfighi et al (6) reported 42 cases of ganglion cell
tumors in the posterior pituitary in 1996; of these, 32 cases were
mixed pituitary adenomas with gangliogliomas. Lu and Xu (7) did not find any cases of gangliocytoma
through the pathological analysis of 1,458 cases of sellar region
tumors. Qin and Yan (8) reported a
39-year-old male patient with gangliocytoma in the sellar region in
2004.
Gangliocytomas in the sellar region should be
distinguished from pituitary adenomas. A total of 65% of pituitary
gangliocytomas are accompanied with pituitary adenomas, and there
are no significant differences in their clinical findings. As
gangliocytomas mainly occur in the posterior pituitary lobe,
patients may complain of endocrine symptoms mainly due to
acromegaly and lactation menopause syndrome (2). In the two cases reported in the
present study, the patients had previously been diagnosed with
amenorrhea. One patient complained of acromegaly and their GH level
was marginally elevated.
MRI and CT scanning are the preferred methods for
imaging gangliocytomas (9).
Non-enhanced CT scanning reveals a homogeneous mass with less
attenuation compared with muscle. CT may show calcifications in
two-thirds of cases. Calcification is typically fine and speckled,
but may also be coarse (10,11).
MRI is the modality of choice for evaluating the extension of the
lesion. Gangliocytomas appear homogeneous on MRI and have
relatively intermediate signal intensity on all pulse sequences.
The density, signal and contrast manifestations are similar to
those of a pituitary adenoma, therefore, it is difficult to
distinguish them under MRI and other imaging modalities prior to
surgery (8).
Gangliocytoma is a slowly growing and
non-metastasizing tumor. A biopsy is required to differentiate
gangliocytomas from malignant neuroblastomas, and excision is
usually curative. During the surgery of the present study, it was
found that the texture of the gangliocytoma was significantly more
rigid and tougher than that of the pituitary tumor section, and the
blood supply of the gangliocytoma was moderate. Gangliocytomas, as
fully differentiated neoplasms, do not have the capability to
metastasize, thus extensive surgical resections or chemotherapy are
not typically required (12). The
therapeutic effects are satisfied by surgery alone; recurrence is
predominantly caused by the subtotal resection of the tumor and any
clinical symptoms can be markedly improved by repeating the
surgery.
In the present study, the gross pathology of the
gangliocytoma specimens showed that the tumor had a firm texture
with clear borders, and certain sections were accompanied by cystic
lesions and calcification.
Spectroscopic examinations can reveal that under the
eosinophilic glial background, differently sized and shaped
gangliocytes exhibit irregular distribution patterns (mononuclear,
dinuclear or polynuclear) and Nissl bodies in the cytoplasm. The
tumor tissues contain nerve fibers with and without medullary
sheaths, and immunohistochemical analysis reveals positive staining
for NF, NSE, Syn and chromogranin A (13).
Gangliocytomas must be differentiated from hamartoma
(14). Microscopically, a hamartoma
is shown as ectopic nervous tissue of the pituitary, and is
combined with ganglion cells, astrocytes and branch cells to form
nodular prominents. Histologically, it is not a tumor, but the
accrementition of ganglion cells, astrocytes and branch cells,
which have been differentiated maturely. Gangliocytomas also
require differentiation from the normal neurohypophysis, which is
the normal tissue changing, in which spindle-shaped glial cells are
sparse, and there are no metatypical cells, no hemorrhage and
necrosis, and no Rosenthol fibers and eosinophilic bodies.
Geddes (15) et
al hypothesized that the GH-releasing hormone (GHRH) produced
by ganglion cells, which were heterotopic in the intrasellar
region, may stimulate or accelerate the occurrence of the endocrine
activity of the pituitary adenoma. Another possibility is that the
undifferentiated ganglion cells discarded by the neurohypophysis
transformed into tumor cells due to GHRH, which was excessively
produced by the hypothalamus, while the pituitary cells formed the
pituitary adenoma by GHRH stimulation. Another previous study has
argued that the increasing number of hormone-releasing factors
discharged by the hypothalamic neuron growing in the pituitary
stimulate the proliferation of pituitary endocrine cells, while the
ganglion cells form the mixed pituitary tumors, which are
stimulated by the hormone produced by the pituitary (16). Ulm et al (17,18)
argued that the mixed pituitary tumor originates from the
steroidogenic acute regulatory protein-expressing follicular cells
in the anterior pituitary, which are the primitive, pluripotent
stem cells of the adult pituitary, with the capability of
bidirectional differentiation of the adenohypophysis and
neurohypophysis, and thus form tumors.
In summary, the possible genesis theories of the
miscellaneous tumor include the following: i) The stem cell
differentiation theory; ii) the induced theory, a tumor occurs
first and induces another type of tumor occurrence; iii) the
encounter theory, two tumors of different origins occur
respectively and mix together; iv) the metaplasia theory, one tumor
is the primary tumor, while the other is the metaplastic tumor; and
v) the gene theory, tumorigenic factors act on the oncogenes of the
adenohypophysis and neurohypophysis, and activate cell
tumorigenesis synchronously or successively (17,18).
These mixed pituitary tumors have rarely been
reported worldwide and therefore, future surgeries are required to
confirm the pathological diagnosis and that generally, excision is
curative. Since mixed tumors do not have the ability to
metastatsize, the therapeutic effects may be satisfied by surgery
alone. Recurrence is predominantly caused by the subtotal resection
of the tumor and any clinical symptoms may be improved a second
surgery. Therefore, this report may aid neurological surgeons’
understanding of mixed pituitary tumors.
References
|
1
|
Kovacs K and Horvath E: Tumors of the
pituitary gland. Atlas of Tumor Pathology Fascicle 21. (2nd
series). Armed Forces Institute of Pathology; Washington, D.C: pp.
1–269. 1986
|
|
2
|
Cannon TC, Brown HH, Hughes BM, Wenger AN,
Flynn SB and Westfall CT: Orbital gangliocytoma in a patient with
chronic progressive proptosis. Arch Ophthalmol. 122:1712–1714.
2004.
|
|
3
|
Puchner MJ and Hecnnann HD: Intrasellar
pituitary gangliocyto-adenoma presenting with acromegaly: case
report. Neurosurgery. 42:1197–1199. 1998.
|
|
4
|
Albonico G, Pellegrino G, Maisano M and
Kardon DE: Gangliocytoma of parapharyngeal region. Arch Pathol Lab
Med. 125:1217–1218. 2001.
|
|
5
|
Greenfield JG: The pathological
examination of forty intracranial neoplasms. Brain. 42:29–85.
1919.
|
|
6
|
Towfighi J, Salam MM, Mclendon RE, et al:
Ganglion cell-containing tumors of the pituitary gland. Arch Pathol
Lab Med. 120:369–377. 1996.
|
|
7
|
Lu DH and Xu QZ: Pathological analysis on
1458 cases of intracranial sellar region tumor. Chin J Oncol.
10:205–208. 1998.(In Chinese).
|
|
8
|
Qin JX, Yan XL and Kong FM: Report of one
case of gangliocytoma of the sellar region. Zhongguo Xiandai
Shenjing Jibing Zazhi. 1:63–64. 2004.(In Chinese).
|
|
9
|
Lonergan GJ, Schwab CM, Suarez ES and
Carlson CL: Neuroblastoma, ganglioneuroblastoma and gangliocytoma:
radiologic-pathologic correlation. Radiographics. 22:911–934.
2002.
|
|
10
|
Ichikawa T, Ohtomo K, Araki T, et al:
Gangliocytoma: computed tomography and magnetic resonance features.
Br J Radiol. 69:114–121. 1996.
|
|
11
|
Johnson GL, Hruban RH, Marshall FF and
Fishman EK: Primary adrenal gangliocytoma: CT findings in four
patients. AJR Am J Roentgenol. 169:169–171. 1997.
|
|
12
|
Mclendon RE, Bidgner DD and Bidgner SH:
The lesions encountered in children and young adults. Pathology of
Tumors of the Central Nervous System. 1st edition. Oxford
University Press; New York: pp. 209–216. 2000
|
|
13
|
Iwase T, Nishizawa S, Baba S, et al:
Intrasellar neuronal choristoma associated with growth
hormone-producing pituitary adenoma containing amyloid deposits.
Hum Pathol. 26:925–928. 1995.
|
|
14
|
Kamel OW, Horoupian DS and Silvertmrg GD:
Mixed gangliocytoma-adenoma: a distinct neuroendocrine tumor of the
pituitary fossa. Hum Pathol. 20:1198–1203. 1989.
|
|
15
|
Geddes JF, Jansen CH, Robinson SF, et al:
‘Gangliocytomas’ of the pituitary: a heterogeneous group of lesions
with differing histogenesis. Am J Surg Pathol. 24:607–613.
2000.
|
|
16
|
Russel DS and Rubinstein LJ: Tumors of
central neuroepithelial origin gangliocytomas (gangliocytomas) and
gangliogliomas. Pathology of Tumors of the Nervous System. 5th
edition. Edward Arnold; London: pp. 289–306. 1989
|
|
17
|
Ulm AJ, Yachnis AT, Brat DJ and Rhoton AL
Jr: Pituicytoma: report of two cases and clues regarding
histogenesis. Neurosurgery. 54:753–757. 2004.
|
|
18
|
Inoue K, Couch EF, Takano K and Ogawa S:
The structure and function of follicular-stellar cells in the
anterior pituitary gland. Arch Histol Cytol. 62:205–218. 1999.
|