1. Introduction
The oncostatic effects of melatonin are particularly
relevant to hormone-dependent tumors (1–5). Of
these neoplasias, the most deeply studied have been mammary
adenocarcinomas. Based on the role of the pineal gland in
inhibiting gonadal maturation and sex hormone secretion in mammals,
Cohen et al (1978) introduced the hypothesis that a decrease
in pineal function decreases melatonin levels and induces a
relative ‘hyperestrogenism’, which underlies the development of
breast cancer (6). Since then,
there has been evidence supporting the theory that the antitumor
actions of melatonin in hormone-dependent tumors are mainly based
on the antiestrogenic properties of melatonin (5,7).
The oncostatic effects of melatonin in
hormone-dependent breast cancer were firstly explained by indirect
neuroendocrine mechanisms, such as the downregulation of the
neuroendocrine reproductive axis by melatonin, and the consequent
reduction of estrogenic hormones responsible for the normal and
pathological growth of the mammary gland (8). In addition, it has also been
demonstrated that melatonin may directly interfere with the
activation of the estrogen receptor and counteract the effects of
estrogens at the tumor cell level, thus behaving as a selective
estrogen receptor modulator (7,9–11). In
more recent years, a third neuroendocrine mechanism has been
described in which melatonin is able to reduce the
estrogen-mediated development of breast cancer, involving the
regulation of certain enzymes responsible for the local synthesis
of estrogens, thus behaving as a selective estrogen enzyme
modulator (12–15).
2. Local synthesis of estrogens in breast
cancer epithelial cells and melatonin
The intratumoral metabolism and synthesis of
estrogens, as a result of the interactions of various enzymes, is
considered to play an important role in the pathogenesis and
development of hormone-dependent breast carcinoma (16–19).
In breast cancer, particularly that of postmenopausal women,
estrogens are synthesized in the mammary tissue by transformation
either from androgen precursors, mainly of adrenal origin, or from
biologically inactive estrogens. Breast carcinoma epithelial cells
contain all the enzymes necessary for the local synthesis of
estrogens (Fig. 1). One of the
major pathways involved in the synthesis of estrogens in breast
cancer cells is the aromatase pathway, which transforms androgens
into estrogens (20). Aromatase
activity and expression is markedly higher in breast cancer tissue
than in normal mammary tissue (21,22).
The second pathway involved in estrogen formation is the sulfatase
pathway, which converts estrogen sulfates into estrone and
estradiol (18,19,22).
The final step of steroidogenesis in peripheral tissues is the
conversion of the weak estrone to the potent biologically active
estradiol by the action of the 17β-hydroxysteroid dehydrogenase
activity type 1 (17β-HSD1) (18,19).
In breast cancer tissue, estrogen sulfotransferase is also present,
which converts estrogens into estrogen sulfates. Since the
sulfo-conjugated estrogens are the biologically inactive forms of
the estrogens, another possible way to control the tissular
concentration of active estradiol is to identify new ways to
stimulate the enzymes involved in the sulfate formation (19,22).
In normal breast tissue, there is a high
concentration of circulating inactive steroids, which are the major
precursor substrates of local estrogen production, mainly estrone.
In this tissue, the estrogen sulfotransferase activity and
expression, the enzyme that inactivates estrone and 17β-estradiol,
tend to be increased. However, in breast carcinoma tissue,
aromatase (which converts androgens into estrogens), sulfatase
(which hydrolyzes the estrone sulfates to estrone) and 17β-HSD1
(which converts the estrone to the potent 17β-estradiol) tend to be
overexpressed, whereas the expression of estrogen sulfotransferase
is frequently decreased, which may result in the accumulation of
17β-estradiol in breast cancer tissues (Fig. 2). At the tumor cell level, melatonin
decreases the activity and expression of aromatase, sulfatase and
17β-HSD1, and increases the activity and expression of estrogen
sulfotransferase (12,13,23,24).
Melatonin tends to modify the activity and expression of the
enzymes involved in the local synthesis of estrogens, causing them
to be similar to the expression of enzymes in the mammary normal
tissue, and may thus protect mammary tissue from excessive
estrogenic effects (Fig. 2).
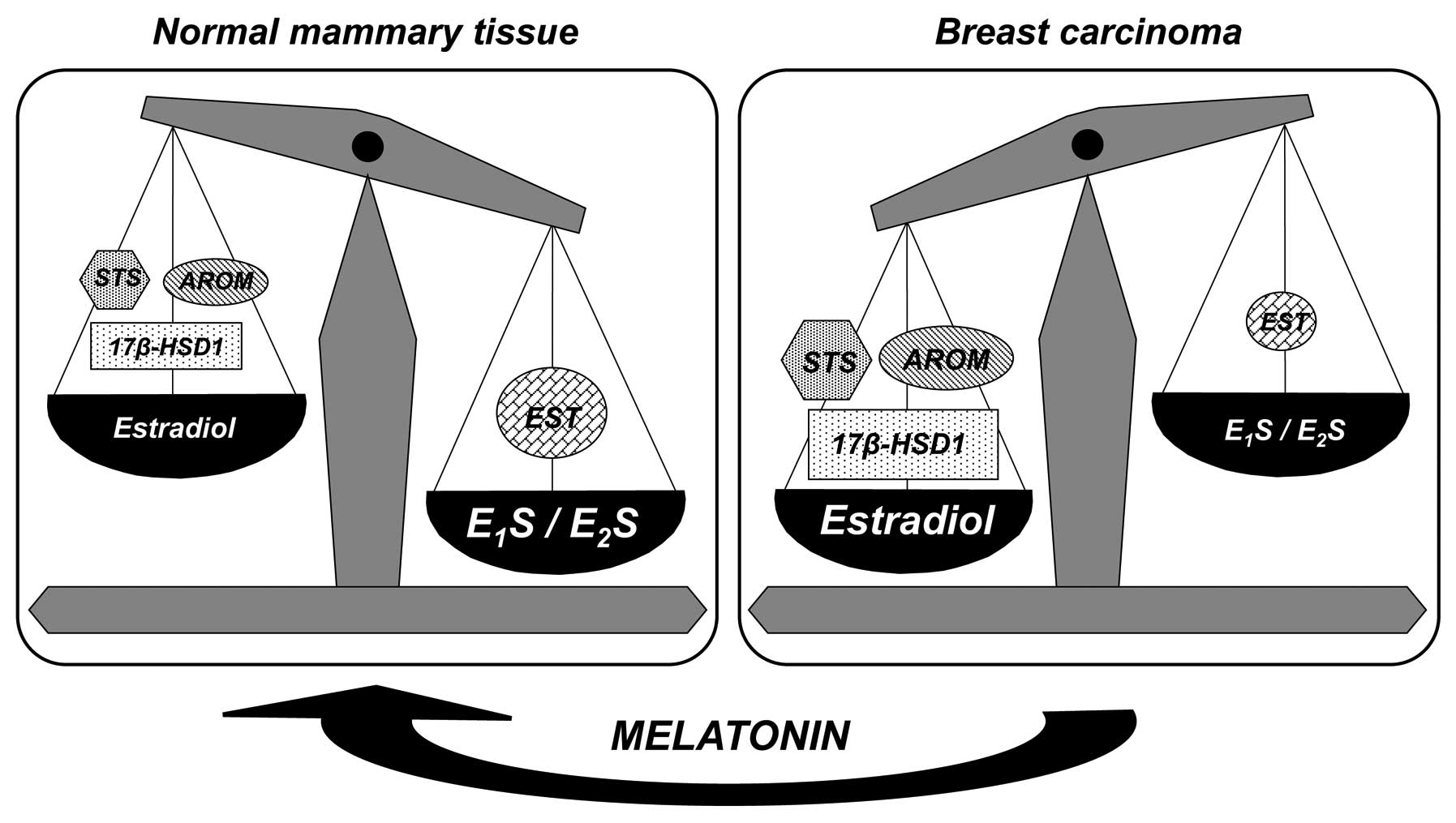 | Figure 2Expression of enzymes associated with
the local production of estrogens in human normal mammary tissue
and breast carcinoma tissue. In the breast carcinoma tissue, STS,
AROM and 17β-HSD1 tend to be overexpressed, while EST is decreased.
Melatonin decreases the expression of AROM, STS and 17β-HSD1, and
increases the EST expression. Thus, melatonin tends to modify the
expression of the enzymes involved in the local synthesis of
estrogens, causing it to be similar to the expression of enzymes in
the mammary normal tissue. Figure modified from Cos et al
(23). STS, sulfatase; AROM,
aromatase; 17β-HSD1, 17β-hydroxysteroid dehydrogenase type 1; EST,
estrogen sulfotransferase; E1S, estrone sulfate;
E2S, estradiol sulfate. |
Regulation of aromatase expression in human tissues
is relatively complex, involving alternative promoter sites that
provide tissue-specific control. In the normal breast, the mammary
adipose tissue maintains low levels of aromatase expression almost
exclusively via promoter I.4 (25).
However, in mammary cancer, both in malignant epithelial cells and
fibroblasts, the expression of aromatase is increased via
activation of promoters II and I.3, which are regulated by cyclic
adenosine monophosphate (cAMP) and factors that regulate cAMP
levels (26). Prostaglandin
E2 (PGE2) is an important regulator of
aromatase gene expression via promoters II and I.3 (25–28).
The formation of PGE2 occurs through the activity of the
cyclooxygenases (COXs), rate-limiting enzymes that catalyze the
conversion of arachidonic acid to prostaglandins. Promoter I.3 and
II are considered to be the major promoters driving aromatase
expression in breast cancer and surrounding adipose tissue. One of
the mechanisms through which melatonin modulates aromatase enzyme
in breast tumor cells is through its downregulatory action on the
expression of COX enzymes, COX-1 and COX-2, which decrease the
levels of PGE2. Lower levels of PGE2 result
in decreased intracellular levels of cAMP, which in turn diminish
the activation of promoters I.3 and II, and result in decreased
aromatase expression (29,30).
In addition, the antiaromatase and antisulfatase
effects of melatonin have also been shown in cancer cell types
other than breast cancer cells. In glioblastoma cells, which
express estrogen receptors and have the ability to synthesize
estrogens, melatonin also reduces the local production of estrogens
by decreasing the activity of aromatase, sulfatase and 17β-HSD1,
and downregulating aromatase, sulfatase and 17β-HSD1 mRNA steady
state levels (31,32).
This melatonin modulatory effect on the aromatase
and sulfatase enzymes, at the tumor cell level, has also been
described in vivo, in rats bearing
7,12-dimethylbenzanthracene-induced mammary tumors. The growth of
these mammary tumors is estrogen-dependent and ovariectomy
significantly reduces both the size and number of tumors, while the
administration of testosterone or estrone sulfate to ovariectomized
animals is able to maintain the tumor growth at the same level as
the control (uncastrated) animals. The stimulatory effects of tumor
development induced by testosterone (which depends on the local
synthesis of estrogens from androgens), due to the aromatase
action, or estrone sulfate (which depends on the estrogens locally
formed by the action of the sulfatase enzyme on the biologically
inactive estrogens), are suppressed by the administration of
melatonin. Tumors from animals treated with melatonin have the
lowest microsomal aromatase and sulfatase activity (14,33).
3. Local synthesis of estrogens in
peritumoral fibroblasts and melatonin
In breast tumors, the majority of aromatase and
sulfatase activity and expression, the two principal pathways of
synthesis of estrogens, are found in the fibroblast component of
the adipose tissue and in vascular endothelial cells. The local
biosynthesis of estrogens in breast cancer depends on paracrine
interactions between malignant epithelial cells and proximal
fibroblasts and vascular endothelial cells. Malignant epithelial
cells secrete cytokines, including tumor necrosis factor α (TNF-α),
interleukin 6 (IL-6) and IL-11, which are upregulated by estrogens.
These cytokines inhibit the differentiation of surrounding
fibroblasts into mature adipocytes, through the selective
inhibition of expression of peroxisome proliferator-activated
receptor γ (PPARγ) and CCAAT/enhancer binding protein α (C/EBPα),
and also stimulate aromatase expression in these undifferentiated
fibroblasts (Fig. 3) (19,34,35).
This biological phenomenon is commonly known as the desmoplastic
reaction or the accumulation of undifferentiated fibroblasts with
high aromatase activity surrounding malignant epithelial cells.
Tumor cells also secrete other factors, such as PGE2,
which stimulate aromatase activity and expression in these
undifferentiated fibroblasts, as well as upregulating
antiadipogenic cytokines.
 | Figure 3Epithelial-stromal interactions in
breast tumors inhibit adipogenic differentiation and enhance
estrogen formation by increasing the aromatase activity of the
undifferentiated fibroblasts. All these actions are mediated by
cytokines, such as TNF-α, IL-6 and IL-11, produced by malignant
epithelial cells. Melatonin reduces the formation of
undifferentiated fibroblasts surrounding malignant epithelial cells
by stimulating the differentiation of fibroblasts to mature
adipocytes and adipogenesis, and by decreasing the aromatase
activity of the fibroblasts and adipocytes through a downregulatory
action on the expression of antiadipogenic cytokines, which
decreases the levels of these cytokines. Lower levels of TNF-α,
IL-6 and IL-11 stimulate the differentiation of fibroblasts and
decrease the aromatase activity and expression. Melatonin also
decreases the production of PGE2 by malignant cells,
which upregulates aromatase expression both in the tumor itself and
in the surrounding adipose tissue, and enhances the production of
IL-11 by tumor cells. Figure modified from Álvarez-García et
al (39) and Cos et al
(23). TNF-α, tumor necrosis
factor-α; IL, interleukin. |
3T3-L1 is a fibroblast cell line that is initially
fibroblastic but which, under appropriate conditions,
differentiates into adipocytes (36). Melatonin treatment during the
preadipocyte differentiation enhances the adipogenesis, and higher
doses of melatonin induce more extensive deposits of lipid droplets
and also induce a ~50% reduction in the aromatase activity of the
cells, two indicators of adipogenic differentiation. It has been
demonstrated that melatonin significantly increases the expression
of PPARγ and C/EBPα, the two main regulators of terminal
adipogenesis (37). An approach to
simulate in vitro the situation occurring in the mammary
tumor is to use cocultures of malignant epithelial cells with
fibroblasts or endothelial cells. The presence of malignant
epithelial cells in the cocultures inhibits the differentiation of
preadipocytes to adipocytes and reduces the intracytoplasmic
triglyceride accumulation, an indicator of adipogenic
differentiation. The presence of malignant cells also stimulates
the aromatase activity in the fibroblasts. Melatonin counteracts
the inhibitory effect on adipocyte differentiation induced by
malignant epithelial cells, and also counteracts the stimulatory
effect of the presence of breast cancer cells on aromatase activity
in fibroblasts (37–39).
The levels of antiadipogenic cytokines, TNF-α, IL-6
and IL-11, in the coculture media are 10-fold higher than those
found in the culture of fibroblasts alone, since epithelial
malignant cells, in the presence of fibroblasts, secrete these
cytokines with the aim to inhibit the differentiation of
preadipocytes into adipocytes and to accumulate undifferentiated
fibroblasts with high aromatase activity around malignant
epithelial cells. The addition of melatonin to the cocultures
decreases the concentrations of cytokines in the media and
counteracts the stimulatory effect induced by the presence of
malignant cells on the cytokines levels. Melatonin also induces a
reduction in the TNF-α, IL-6 and IL-11 mRNA expression in breast
cancer epithelial cells and fibroblasts (Fig. 3). The addition of luzindole, a
melatonin receptor antagonist, prevents this inhibitory effect of
melatonin on cytokines expression, indicating that melatonin acts
through known melatonin receptor-mediated mechanisms (39).
In summary, melatonin may reduce the level of
undifferentiated fibroblasts surrounding malignant epithelial cells
by stimulating the differentiation of fibroblasts to mature
adipocytes and adipogenesis, and by decreasing the aromatase
activity of the fibroblasts through a downregulatory action on the
expression of antiadipogenic cytokines, which decreases the levels
of these cytokines. Lower levels of TNF-α, IL-6 and IL-11 allow the
differentiation of fibroblasts, as well as decreasing the aromatase
activity and expression. Melatonin also decreases the production of
PGE2 by malignant cells, which downregulates aromatase
expression and cytokine production in the tumor itself and in the
surrounding adipose tissue. Lower levels of aromatase lead to lower
levels of estrogens, resulting in decreased growth and development
of the breast tumor (Fig. 3).
4. Local synthesis of estrogens in
peritumoral endothelial cells and melatonin
Endothelial cells also represent a critical cellular
element in the tumor microenvironment, which play a crucial role in
the growth and progression of breast tumors. They are another
source of estrogens, as they also express aromatase (40,41).
Promoter I.7 is a novel breast cancer-associated aromatase promoter
mainly active in vascular endothelial cells, and is upregulated in
breast cancer tissue (42).
Excessive aromatase expression via promoters I.3, II and I.7, and
consequent increase in estrogen biosynthesis in malignant
epithelial cells, undifferentiated adipose fibroblasts and adjacent
endothelial cells contribute to the development and progression of
breast cancer. In addition, endothelial cells provide structural
and biochemical support for tumor growth and progression of cancer
through control of angiogenesis. Vascular endothelial growth factor
(VEGF) secreted by breast cancer cells is essential for the
expansion of breast cancer and may function in both paracrine and
autocrine manners to promote the proliferation, growth, survival
and migration of endothelial cells (43,44).
In endothelial cells, melatonin decreases the
aromatase activity and expression mainly by inducing a significant
downregulation in aromatase expression specifically driven by
promoter I.7, the major promoter directing aromatase expression in
endothelial cells (Fig. 4)
(45).
VEGF, a major regulator of endothelial growth, added
to endothelial cell cultures stimulates the proliferation of these
cells and melatonin counteracts this effect (46). Melatonin reduces VEGF mRNA
expression in human breast cancer (MCF-7) cells and also reduces
VEGF levels in cell culture media of malignant epithelial cells
(Fig. 4). Cocultures of breast
malignant epithelial cells and endothelial cells is an approach to
simulate in vitro the paracrine interaction between these
cells in the mammary tumors. The presence of malignant epithelial
cells in the cocultures is able to stimulate the endothelial cell
proliferation and increase the VEGF levels in the culture media.
Melatonin counteracts the stimulatory effects on endothelial cell
proliferation and on VEGF protein levels in the coculture media.
The changes in endothelial cell proliferation induced by melatonin
are mediated by an inhibition of the synthesis of VEGF in malignant
epithelial cells. Conditioned media from malignant cells stimulate
endothelial cell proliferation, and this effect is significantly
counteracted by anti-VEGF and melatonin (46).
All these findings suggest that melatonin may play a
role in the paracrine interactions between malignant epithelial
cells and proximal endothelial cells, through a downregulatory
action on VEGF expression in human breast cancer cells, which
decreases the levels of VEGF surrounding endothelial cells. Lower
levels of VEGF may be important in reducing the number of
estrogen-producing cells proximal to malignant cells, as well as in
decreasing tumoral angiogenesis. Antiangiogenic activity of
melatonin against the pro-angiogenic effects of breast cancer cells
has also been described (47).
Recently, it has been demonstrated that melatonin has effects on
different steps of the angiogenic process in endothelial cell
cultures (47). Melatonin strongly
inhibits the proliferation of endothelial cells and counteracts the
stimulatory effect induced by estradiol. In Transwell assays,
melatonin has been identified to reduce the number of endothelial
cells that invaded through a basement membrane in response to VEGF.
Endothelial cell migration is essential for the formation of new
blood vessels during neo-angiogenesis. Melatonin treatment strongly
inhibits the migration of endothelial cells in wound-healing
assays. Another important step during neo-angiogenesis is the
formation of tubes by endothelial cells. It is established that
VEGF increases the formation of a branching network of tubes.
Melatonin disrupts the tube formation and counteracts the
VEGF-stimulated tubular network formation by endothelial cells. In
addition, conditioned media collected from breast cancer cells are
angiogenically active and stimulate tubule length formation. This
effect is significantly counteracted by the addition of either
anti-VEGF antibody or melatonin, which suggests that the
melatonin-induced decrease of capillary structure formation
stimulated by conditioned media from MCF-7 cells may occur as a
result of inhibition of VEGF activity (47).
Melatonin may play a role in the paracrine
interactions that take place between malignant epithelial cells and
proximal endothelial cells, acting by different mechanisms. On one
hand, melatonin exerts antiangiogenic effects and may be important
in reducing endothelial cell proliferation, invasion, migration and
tube formation, through a downregulatory action on VEGF and
PGE2 (Fig. 4).
PGE2 synthesis induced by VEGF may directly promote
angiogenesis and melatonin through its downregulatory action on the
expression of COX enzymes, which decrease the levels of
PGE2 and reduce angiogenesis. On the other hand,
melatonin inhibits aromatase activity and expression in endothelial
cells by regulating gene expression of specific aromatase promoter
regions, thereby reducing the local production of estrogens
(Fig. 4).
5. Conclusions
Several lines of evidence highlight the contribution
of the tumor microenvironment to its growth and maintenance. Cells
immediately adjacent to the tumor are not only passive structural
support but also active elements in tumor progression. Among the
numerous different cell types surrounding breast cancer cells, the
most abundant are those that compose mammary adipose tissue. Ninety
percent of these resident cells of adipose tissue are fibroblasts,
the precursors of mature adipocytes, and 7% are endothelial cells
(35). Epithelial-stromal
interactions in breast tumors inhibit adipogenic differentiation
and enhance estrogen formation by increasing the aromatase activity
of the undifferentiated fibroblasts. All these actions are mediated
by cytokines, such as TNF-α, IL-11 and IL-6, produced by malignant
epithelial cells. Melatonin may reduce the formation of
undifferentiated fibroblasts surrounding malignant epithelial cells
by stimulating the differentiation of fibroblasts to mature
adipocytes and adipogenesis, and by decreasing the aromatase
activity of the fibroblasts and adipocytes through a downregulatory
action on the expression of antiadipogenic cytokines, which
decrease the levels of these cytokines. Lower levels of TNF-α, IL-6
and IL-11 stimulate the differentiation of fibroblasts and decrease
the aromatase activity and expression. Melatonin also decreases the
production of PGE2 by malignant cells, which upregulates
aromatase expression both in the tumor itself and in the
surrounding adipose tissue and enhances the production of IL-11 by
tumor cells. Endothelial cells also produce estrogens from
androgens precursors. Melatonin decreases the activation of
promoter I.7 and results in decreased aromatase expression. In
addition, melatonin reduces endothelial cell proliferation,
invasion, migration and tube formation, through a downregulatory
action on VEGF. This melatonin modulation of epithelial-stromal
interactions favors lower numbers of undifferentiated fibroblasts,
angiogenesis and reduced local estrogen concentrations in breast
tumors.
Melatonin may play a role in the paracrine
interactions that occur between malignant epithelial cells and
proximal adipose and endothelial cells, through a downregulatory
action on cytokines and growth factors produced by breast tumor
cells. The actions of melatonin described in the present review
involve antiproliferative, antiaromatase and antiangiogenic
effects, and suggest that melatonin may potentially be beneficial
as an anticancer drug in the prevention and treatment of
estrogen-dependent mammary tumors. Therefore, this creates
interesting possibilities for the clinical applications of
melatonin in breast cancer.
Acknowledgements
This study was supported by grants from the Spanish
Ministry of Science and Innovation (SAF2010-19579) and the
Valdecilla Research Institute (APG-09-GC6).
References
|
1
|
Cos S and Sánchez-Barceló EJ: Melatonin
and mammary pathological growth. Front Neuroendocrinol. 21:133–170.
2000.
|
|
2
|
Cos S and Sánchez-Barceló EJ: Melatonin,
experimental basis for a possible application in breast cancer
prevention and treatment. Histol Histopathol. 15:637–647. 2000.
|
|
3
|
Blask DE, Sauer LA and Dauchy RT:
Melatonin as a chronobiotic/anticancer agent: cellular, biochemical
and molecular mechanisms of action and their implications for
circadian-based cancer therapy. Curr Topics Med Chem. 2:113–132.
2002.
|
|
4
|
Sánchez-Barceló EJ, Cos S, Fernández R and
Mediavilla MD: Melatonin and mammary cancer: a short review. Endocr
Relat Cancer. 10:153–159. 2003.
|
|
5
|
Sánchez-Barceló EJ, Cos S, Mediavilla MD,
Martínez-Campa CM, González A and Alonso-González C:
Melatonin-estrogen interactions in breast cancer. J Pineal Res.
38:217–222. 2005.
|
|
6
|
Cohen M, Lippman M and Chabner B: Role of
pineal gland in aetiology and treatment of breast cancer. Lancet.
2:814–816. 1978.
|
|
7
|
Cos S, González A, Martínez-Campa C,
Mediavilla MD, Alonso-González C and Sánchez-Barceló EJ:
Estrogen-signaling pathway: a link between breast cancer and
melatonin oncostatic actions. Cancer Detect Prev. 30:118–128.
2006.
|
|
8
|
Reiter RJ: The pineal and its hormones in
the control of reproduction in mammals. Endocr Rev. 1:109–131.
1980.
|
|
9
|
Molis TM, Spriggs LL and Hill SM:
Modulation of estrogen receptor mRNA expression by melatonin in
MCF-7 human breast cancer cells. Mol Endocrinol. 8:1681–1690.
1994.
|
|
10
|
Cos S, Blask DE, Lemus-Wilson A and Hill
SM: Effects of melatonin on the cell cycle kinetics and estrogen
rescue of MCF-7 human breast cancer cells in culture. J Pineal Res.
10:36–42. 1991.
|
|
11
|
Hill SM, Spriggs LL, Simon MA, Muraoka H
and Blask DE: The growth inhibitory action of melatonin on human
breast cancer cells is linked to the estrogen response system.
Cancer Lett. 64:249–256. 1992.
|
|
12
|
Cos S, Martínez-Campa C, Mediavilla MD and
Sánchez-Barceló EJ: Melatonin modulates aromatase activity in MCF-7
human breast cancer cells. J Pineal Res. 38:136–142. 2005.
|
|
13
|
González A, Martínez-Campa C, Mediavilla
MD, et al: Effects of MT1 melatonin receptor overexpression on the
aromatase-suppressive effect of melatonin in MCF-7 human breast
cancer cells. Oncol Rep. 17:947–955. 2007.
|
|
14
|
Cos S, González A, Güezmes A, Mediavilla
MD, Martínez-Campa C, Alonso-González C and Sánchez-Barceló EJ:
Melatonin inhibits the growth of DMBA-induced mammary tumors by
decreasing the local biosynthesis of estrogens through the
modulation of aromatase activity. Int J Cancer. 118:274–278.
2006.
|
|
15
|
Martínez-Campa C, González A, Mediavilla
MD, Alonso-González C, Sánchez-Barceló EJ and Cos S: Melatonin
enhances the inhibitory effect of aminoglutethimide on aromatase
activity in MCF-7 human breast cancer cells. Breast Cancer Res
Treat. 94:249–254. 2005.
|
|
16
|
Simpson ER: Role of aromatase in sex
steroid action. J Mol Endocrinol. 25:149–156. 2000.
|
|
17
|
van Landeghem AA, Poortman J, Nabuurs M
and Thijssen JH: Endogenous concentration and subcellular
distribution of estrogens in normal and malignant human breast
tissue. Cancer Res. 45:2900–2906. 1985.
|
|
18
|
Pasqualini JR: The selective estrogen
enzyme modulators in breast cancer: a review. Biochim Biophys Acta.
1654:123–143. 2004.
|
|
19
|
Pasqualini JR and Chetrite GS: Recent
insight on the control of enzymes involved in estrogen formation
and transformation in human breast cancer. J Steroid Biochem Mol
Biol. 93:221–236. 2005.
|
|
20
|
Conley A and Hinshelwood M: Mammalian
aromatases. Reproduction. 121:685–695. 2001.
|
|
21
|
Santen RJ and Harvey HA: Use of aromatase
inhibitors in breast carcinoma. Endocr Relat Cancer. 6:75–92.
1999.
|
|
22
|
Suzuki T, Miki Y, Nakamura Y, et al: Sex
steroid-producing enzymes in human breast cancer. Endocr Relat
Cancer. 12:701–720. 2005.
|
|
23
|
Cos S, González A, Álvarez-García V,
Alonso-González C and Martínez-Campa C: Melatonin and breast
cancer: selective estrogen enzyme modulator. Advances in Cancer
Drug Targets. Atta-ur-Rahman: 1. 1st Edition. Bentham Science
Publishers; Sharjah (UAE): pp. 207–237. 2012
|
|
24
|
González A, Cos S, Martínez-Campa C,
Alonso-González C, Sánchez-Mateos S, Mediavilla MD and
Sánchez-Barceló EJ: Selective estrogen enzyme modulator actions of
melatonin in human breast cancer cells. J Pineal Res. 45:86–92.
2008.
|
|
25
|
Bulun SE, Lin Z, Imir G, et al: Regulation
of aromatase expression in estrogen-responsive breast and uterine
disease: from bench to treatment. Pharmacol Rev. 57:359–383.
2005.
|
|
26
|
Bulun SE, Sebastian S, Takayama K, Suzuki
T, Sasano H and Shozu M: The human CYP19 (aromatase p450) gene:
update on physiologic roles and genomic organization of promoters.
J Steroid Biochem Mol Biol. 86:219–224. 2003.
|
|
27
|
Díaz-Cruz ES, Shapiro CL and Brueggemeier
RW: Cyclooxygenase inhibitors suppress aromatase expression and
activity in breast cancer cells. J Clin Endocrinol Metab.
90:2563–2570. 2005.
|
|
28
|
Prosperi JR and Robertson FM:
Cyclooxygenase-2 directly regulates gene expression of P450 Cyp19
aromatase promoter regions pII, pI. 3 and pI7 and estradiol
production in human breast tumor cells. Prostaglandins Other Lipid
Mediat. 81:55–70. 2006.
|
|
29
|
Martínez-Campa C, González A, Mediavilla
MD, Alonso-González, Álvarez-García V, Sánchez-Barceló EJ and Cos
S: Melatonin inhibits aromatase promoter expression by regulating
cyclooxygenases expression and activity in breast cancer cells. Br
J Cancer. 101:1613–1619. 2009.
|
|
30
|
Wang J, Xiao X, Zhang Y, et al:
Simultaneous modulation of COX-2, p300, Akt, and Apaf-1 signaling
by melatonin to inhibit proliferation and induce apoptosis in
breast cancer cells. J Pineal Res. 53:77–90. 2012.
|
|
31
|
González A, Martínez-Campa C, Mediavilla
MD, Alonso-González C, Sánchez-Barceló EJ and Cos S: Inhibitory
effects of pharmacological doses of melatonin on aromatase activity
and expresión in rat glioma cells. Br J Cancer. 97:755–760.
2007.
|
|
32
|
González A, Martínez-Campa C, Mediavilla
MD, Alonso-González C, Álvarez-García V, Sánchez-Barceló EJ and Cos
S: Inhibitory effects of melatonin on sulfatase and
17β-hydroxysteroid dehydrogenase activity and expression in glioma
cells. Oncol Rep. 23:1173–1178. 2010.
|
|
33
|
González A, Álvarez-García V,
Martínez-Campa C, Mediavilla MD, Alonso-González C, Sánchez-Barceló
EJ and Cos S: In vivo inhibition of the estrogen sulfatase enzyme
and growth of DMBA-induced mammary tumors by melatonin. Curr Cancer
Drug Tar. 10:279–286. 2010.
|
|
34
|
Meng L, Zhou J, Sasano H, Suzuki T,
Zeitoun KM and Bulun SE: Tumor necrosis factor α and interleukin 11
secreted by malignant breast epitelial cells inhibit adipocyte
differentiation by selectively down-regulating CCAAT/enhancer
binding protein α and peroxisome proliferator-activated receptor γ:
mechasnism of desmoplastic reaction. Cancer Res. 61:2250–2255.
2001.
|
|
35
|
Bulun SE, Chen D, Lu M, et al: Aromatase
excess in cancers of breast, endometrium and ovary. J Steroid
Biochem Mol Biol. 106:81–96. 2007.
|
|
36
|
Ntambi JM and Kim YC: Adipocyte
differentiation and gene expression. J Nutr. 130:3122S–3126S.
2000.
|
|
37
|
González A, Álvarez-García V,
Martínez-Campa C, Alonso-González C and Cos S: Melatonin promotes
differentiation of 3T3-L1 fibroblasts. J Pineal Res. 52:12–20.
2012.
|
|
38
|
Knower KC, To SQ, Takagi K, et al:
Melatonin suppresses aromatase expression and activity in breast
cancer associated fibroblasts. Breast Cancer Res Treat.
132:765–771. 2012.
|
|
39
|
Álvarez-García V, González A,
Alonso-González C, Martínez-Campa C and Cos S: Melatonin interferes
in the desmoplastic reaction in breast cancer by regulating
cytokine production. J Pineal Res. 52:282–290. 2012.
|
|
40
|
Harada N, Sasano H, Murakami H, Ohkuma T,
Nagura H and Takagi Y: Localized expression of aromatase in human
vascular tissues. Circ Res. 84:1285–1291. 1999.
|
|
41
|
Mukherjee TK, Dinh H, Chaudhuri G and
Nathan L: Testosterone attenuates expression of vascular cell
adhesion molecule-1 by conversion to estradiol by aromatase in
endothelial cells: implications in atherosclerosis. Proc Nat Acad
Sci USA. 6:4055–4060. 2002.
|
|
42
|
Sebastian S, Takayama K, Shozu M and Bulun
SE: Cloning and characterization of a novel endothelial promoter of
the human CYP19 (aromatase P450) gene that is upregulated in breast
cancer tissue. Mol Endocrinol. 10:2243–2254. 2001.
|
|
43
|
Senger DR, Van De Water L, Brown LF, et
al: Vascular permeability factor (VPF, VEGF) in tumor biology.
Cancer Metastasis Rev. 12:303–324. 1993.
|
|
44
|
Liang Y and Hyder SM: Proliferation of
endothelial and tumor epithelial cells by progestin-induced
vascular endothelial growth factor from human breast cancer cells:
paracrine and autocrine effects. Endocrinology. 146:3632–3641.
2005.
|
|
45
|
Álvarez-García V, González A,
Martínez-Campa C, Alonso-González C and Cos S: Melatonin modulates
aromatase activity and expression in endotelial cells. Oncol Rep.
29:2058–2064. 2013.
|
|
46
|
Álvarez-García V, González A,
Alonso-González C, Martínez-Campa C and Cos S: Regulation of
vascular endotelial growth factor by melatonin in human breast
cáncer cells. J Pineal Res. 54:373–380. 2013.
|
|
47
|
Álvarez-García V, González A,
Alonso-González C, Martínez-Campa C and Cos S: Antiangiogenic
effects of melatonin in endotelial cell cultures. Microvasc Res.
87:25–33. 2013.
|
















