Introduction
Cholangiocarcinoma, an aggressive malignant tumor
that develops from the bile duct epithelium, is associated with
local invasiveness and a high rate of metastasis (1,2). The
worldwide incidence and mortality rates associated with
cholangiocarcinoma have risen over the past three decades. In
Thailand, the annual incidence of cholangiocarcinoma is 87 per
100,000 inhabitants (3). In the
United States, the most commonly recognized risk factor for
cholangiocarcinoma is primary sclerosing cholangitis (4). However, in Southeast Asia and
particularly in Thailand, infection with hepatobiliary flukes
(Opisthorchis viverrini) is the most common risk factor for
cholangiocarcinoma (5). Therapeutic
options for cholangiocarcinoma patients are limited, as this type
of cancer responds poorly to chemotherapy and radiation therapy.
Surgery is thus the only potentially effective treatment for
cholangiocarcinoma. However, typical five-year survival rates of
32–50% are achieved only by a small number of patients with
negative histological margins at the time of surgery (6–8).
Therefore, the understanding of the mechanisms involved in cancer
cell invasion and metastasis may be useful in developing new
therapeutic options for cholangiocarcinoma patients.
The function of the extracellular matrix (ECM) in
the tumor microenvironment is not limited to forming a barrier
against tumor invasion. Previous studies have indicated that
interactions between cancer cells and the ECM play an important
role in cancer progression. The molecular components of the ECM,
such as fibronectin, laminin, collagen and heparin sulfate
proteoglycans, communicate with cancer cells and modulate a variety
of cellular functions required for cancer cells to exhibit invasive
and metastatic properties (9–11).
Numerous results from pathological studies have indicated that
cholangiocarcinoma cells are surrounded by a dense sheath of
connective tissue that contains the ECM (12–14).
However, there have been no studies to date regarding the
definitive role that the ECM plays in cholangiocarcinoma cell
invasion. Therefore, we aimed to investigate the involvement of the
ECM in cholangiocarcinoma cell invasion.
Materials and methods
Cell cultures
The RMCCA1 human cholangiocarcinoma cell line,
originally derived from a cholangiocarcinoma patient (15), was grown in Ham’s F12 medium (Gibco,
Grand Island, NY, USA) supplemented with 10% fetal bovine serum
(Gibco) at 37°C in a 5% CO2 humidified atmosphere.
Cell invasion assay
To study the mechanism of cancer cell invasion in
vitro, RMCCA1 cells were cultured in BD Matrigel matrix (BD
Biosciences, Bedford, MA, USA) for 0–24 h. Next, cancer cells were
seeded into porous cell culture insert cups (BD Biosciences) each
containing a layer of matrix gel. The number of cancer cells that
invaded through the basement membrane within 24 h was assessed by
staining the cells with crystal violet (Sigma-Aldric, St. Louis,
MO, USA) (16).
Two-dimensional (2D) gel
electrophoresis
2D gel electrophoresis was performed for the
analysis of proteins extracted from cholangiocarcinoma cells
cultured in uncoated and 24-h matrix gel-coated plates. Each
electrophoresis gel contained three pooled samples from the cell
culture plates. Six gels were prepared in biological triplicates
from the uncoated and matrix gel-coated plates. Protein samples
(500 μg) were applied to 18-cm immobilized pH gradient (IPG) gel
strips (pH 3–10; GE Healthcare, Uppsala, Sweden) by cup loading
near the anodic ends of the strips. Isoelectric focusing (IEF) was
performed using an Ettan IPGphor Manifold on an Ettan IPGphor
isoelectric focusing unit (GE Healthcare) for 32,000 Vh at 20°C.
Following IEF, each gel strip was equilibrated with equilibration
buffer. The IPG strips were then loaded and run on 12.5% acrylamide
gels (GE Healthcare) using the Ettan DALTsix electrophoresis system
(GE Healthcare). The run was stopped after the bromophenol blue dye
front had run off the bottom of the gels. The gels were then
stained with colloidal Coomassie Blue (GE Healthcare).
2D image analysis
The proteins were visualized using an ImageScanner
(GE Healthcare). The gel images were analyzed to determine
differential protein expression profiles using ImageMaster 2D
Platinum software (GE Healthcare). Student’s t-test was used for
statistical analysis and P<0.05 was considered to indicate a
statistically significant difference.
Protein identification by liquid
chromatography-tandem mass spectrometry (LC-MS/MS)
In-gel digestion
LC-MS/MS was performed by the Proteomics Laboratory,
Genome Institute, National Science and Technology Development
Agency (Pathumthani, Thailand). Following 2D analysis, an in-gel
digestion was performed. Briefly, after the protein spots were
excised, the gel plugs were dehydrated with 100% acetonitrile
(ACN), reduced with 10 mM DTT in 10 mM ammonium bicarbonate at room
temperature for 1 h and alkylated at room temperature for 1 h in
the dark in the presence of 100 mM iodoacetamide in 10 mM ammonium
bicarbonate. Following alkylation, the gel pieces were dehydrated
twice with 100% ACN for 5 min. For the in-gel digestion of the
proteins, 10 μl trypsin solution (20 ng/μl trypsin in 50% ACN/10 mM
ammonium bicarbonate) was added to the gels followed by incubation
at room temperature for 20 min. Next, 20 μl 30% ACN was added to
keep the gels immersed throughout digestion. The gels were
incubated at 37°C overnight. To extract the peptide digestion
products, 30 μl 50% ACN in 0.1% formic acid was added to the gels,
which were then incubated at room temperature for 10 min in a
shaker. The extracted peptides were collected and pooled in a new
tube. The pooled extracted peptides were dried by vacuum
centrifugation at 2,500 × g for 10 min and stored at −80°C until
further mass spectrometric analysis.
LC-MS/MS analysis
The LC-MS/MS analysis of the digested peptide
mixtures was performed using a Waters SYNAPT™ HDMS™ system (Waters,
Milford, MA, USA). The 1D-nanoLC was performed with a Waters
nanoACQUITY UPLC system (Waters). Tryptic digests (4 μl) were
injected onto an reversed-phase analytical column (20 cm × 75 μm)
packed with 1.7-μm ethylene bridged hybrid C18 material (Waters).
The peptides were eluted with a linear gradient of 2–40%
acetonitrile developed over 30 min at a flow rate of 1000 nl/min.
This elution was followed by a 10-min 80% acetonitrile treatment to
clean the column before using 2% acetonitrile for the next sample.
The effluent samples were electrosprayed into a mass spectrometer
(SYNAPT HDMS system) for MS/MS analysis of the peptides, and
spectral data were generated for further protein identification by
matching against hits in a database search.
Mass lists in the form of Mascot generic files were
created and used as the inputs for the Mascot MS/MS Ion web-based
search functionality at the National Center for Biotechnology
Information non-redundant database (www.matrixscience.com). The default search parameters
were applied as follows: Enzyme, trypsin; taxonomy, Homo
sapiens (human); maximum missed cleavages, 1; fixed
modifications, carbamidomethyl (C); variable modifications,
oxidation (M); peptide tolerance, ±1.2 Da; MS/MS tolerance, ±0.6
Da; peptide charge, 1+, 2+ and 3+; and instrument,
ESI-QUAD-TOF.
Western blot analysis
Protein extracts isolated from the cells cultured in
the uncoated and 24-h matrix gel-coated plates were separated by
12% SDS-PAGE and then transferred onto a nitrocellulose membrane
(GE Healthcare). The membrane was subsequently incubated with
monoclonal antibodies against L-plastin (1:50; Santa Cruz
Biotechnology, Santa Cruz, CA, USA) and β-actin (1:500; Cell
Signaling Technology, Danvers, MA, USA). Horseradish
peroxidase-conjugated anti-mouse immunoglobulin G (IgG) and
anti-rabbit IgG at 1:5,000 dilutions were used as secondary
antibodies (GE Healthcare). The blots were visualized using an ECL
Plus detection kit and Hyperfilm ECL (GE Healthcare). The western
blot results were quantified using densitometer and image analysis
software (ImageScanner III and ImageQuant TL; GE Healthcare,
Uppsala, Sweden).
Inhibition of L-plastin expression
using transient siRNA transfection
L-plastin siRNA (Santa Cruz Biotechnology) was used
to knock down L-plastin gene expression. A fluorescein-labeled,
double-stranded RNA duplex (BLOCK-iT™ Fluorescent Oligo;
Invitrogen, Melville, NY, USA) was designed as a control. The siRNA
molecules were diluted in Opti-MEM® I Medium without
serum (Gibco) and mixed gently. Next, Lipofectamine™ 2000
(Invitrogen) was diluted in Opti-MEM I Medium without serum, mixed
gently and incubated for 5 min at room temperature. The diluted
siRNA molecules and diluted Lipofectamine 2000 were then combined.
The mixtures were incubated for 20 min at room temperature to allow
for complex formation to occur. The siRNA molecule-Lipofectamine
2000 complexes were added to each well containing cells and medium,
and mixed gently by rocking the plate back and forth. The cells
were incubated at 37°C in a CO2 incubator for 6 h. Next,
the growth medium was replaced after 6 h, and the cells were
harvested 24 h after transfection. Western blotting analysis using
the L-plastin antibody was performed to assess the degree of
L-plastin gene expression knockdown.
Immunofluorescence microscopy
Cells (5×104) were incubated with the
primary antibody, anti-L-Plastin (1:10), for 1 h at room
temperature. The cells were then washed and incubated with the
appropriate secondary antibody (Alexa Fluor 594, anti-mouse;
Molecular Probes, Grand Island, NY, USA) for 1 h at room
temperature. The actin filaments (F-actin) in the cell cytoplasm
were stained with Alexa Fluor 488 phalloidin (Molecular Probes),
and the nuclei were stained with TOPO3 (Molecular Probes). The
cover slides were removed from the plates and mounted with antifade
on the slides. Cell images were captured with a confocal scanning
biological microscope (FV1000; Olympus, Tokyo, Japan).
Immunohistochemical staining
The study was performed with approval from the
Ethics Committee of Rajavithi Hospital (Bangkok, Thailand).
Paraffinized sections on glass slides were subjected to L-plastin
detection by standard immunohistochemical technique. Sections were
hybridized overnight at 4°C with a 1:50 dilution of L-plastin
antibody (Abcam, Cambridge, MA, USA), followed by incubation with
the secondary antibody, polyclonal Goat Anti-Mouse IgG (Abcam),
conjugated to horseradish peroxidase that catalyzes a
color-producing reaction (Abcam). The signals were visualized under
high power magnification (x200) on an Olympus BH2 microscope.
Statistical analysis
Continuous values for the observed levels in the
invasion assay were expressed as the mean and standard deviation.
One-way analysis of variance was used for the analysis of the
multiple variables of the cell invasion assay. Student’s t-test was
employed to evaluate the mean differences in the intensity volume
of each corresponding spot between the two groups of samples. The
statistical analysis of the immunohistochemical studies was
performed using either the χ2 test or Fisher’s exact
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Culturing cholangiocarcinoma cells in
matrix gel increases their invasiveness
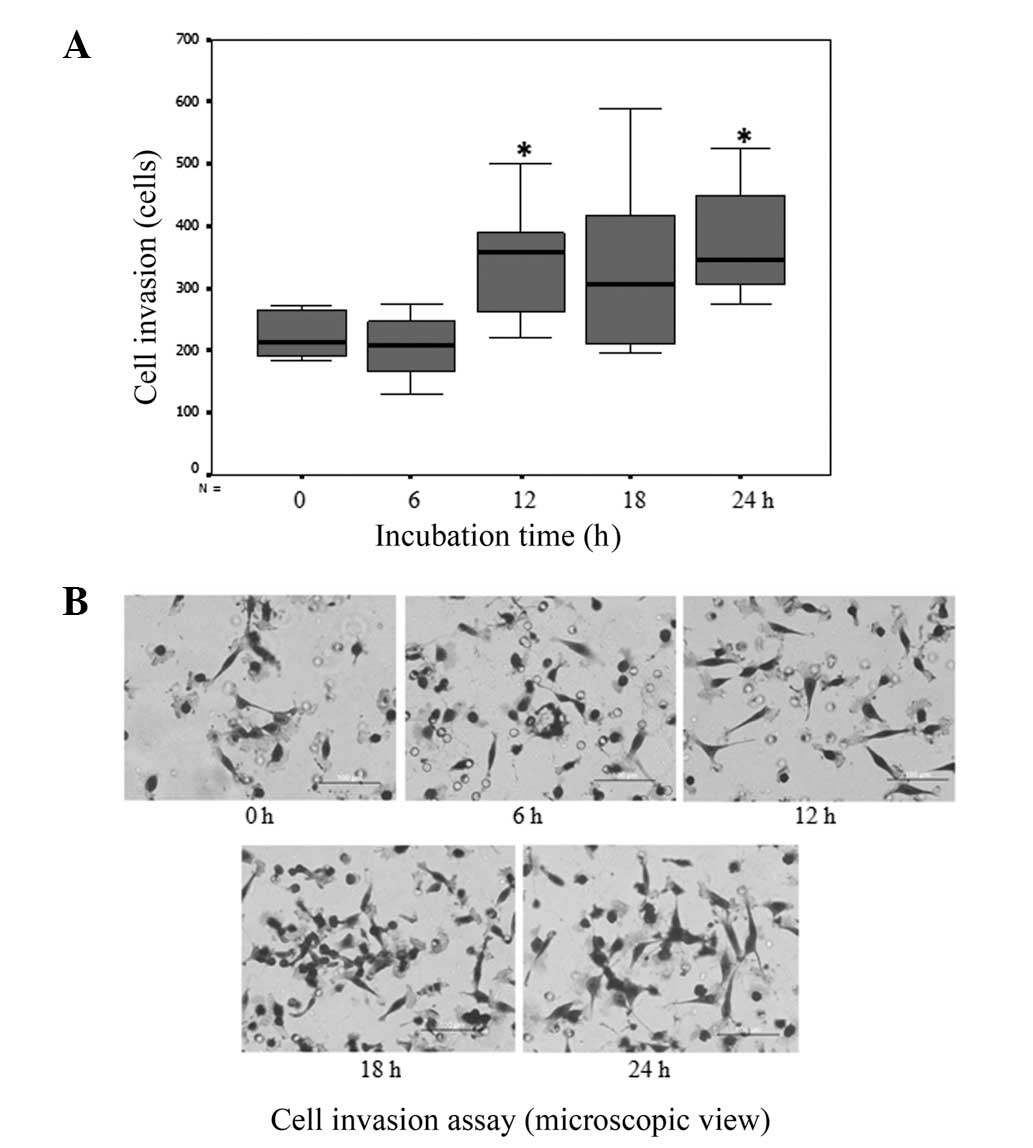
RMCCA1 cholangiocarcinoma cells were incubated in
matrix gel for 0–24 h, and invasion assays were then performed. The
results showed that a significantly higher number of
cholangiocarcinoma cells that were cultured in matrix gel invaded
through the insertion cup compared with that observed with the
cells that were cultured on uncoated plates (P<0.001; Fig. 1).
Proteomic study of cholangiocarcinoma
cells cultured in matrix gel
To investigate the proteins potentially involved in
cholangiocarcinoma cell invasion, cholangiocarcinoma cells were
cultured in plates coated with or without matrix gel. Next, 2D gel
electrophoresis using pH 3–10 Linear IPG strips was performed to
identify the protein expression profiles of these cells.
Approximately 800 protein spots were detected by colloidal
Coomassie staining. Quantitative intensity and statistical analyses
identified 129 protein spots with significantly altered expression
levels in matrix gel culture compared with the uncoated culture
system. Of these 129 proteins, 60 proteins exhibited greater than
two-fold upregulation as determined by mass spectrometry. All the
identified proteins were in the expected ranges of their
theoretical molecular masses and pI values (Table I). We report for the first time that
the ECM plays a major role in the regulation of cholangiocarcinoma
cell invasion. Based on 2D electrophoresis results, we identified
the proteins that were upregulated when cholangiocarcinoma cells
were cultured in matrix gel for 24 h.
 | Table IA summary of upregulated proteins
expressed in cholangiocarcinoma cells cultured in matrix gel, as
identified by Q-TOF MS and MS/MS analyses. |
Table I
A summary of upregulated proteins
expressed in cholangiocarcinoma cells cultured in matrix gel, as
identified by Q-TOF MS and MS/MS analyses.
| Functional category
and protein name | GI number | Mr | pI | Score | Coverage % | Ratio | Gene ID | Cellular
component |
|---|
| Actin-binding
protein |
| L-plastin | 62087548 | 56,196 | 5.21 | 52 | 6 | 4.6 | LCP1, PLS2 | Cytoplasm, cell
membrane, cytoskeleton |
| Cytovillin 2
(Ezrin) | 340217 | 68,233 | 5.80 | 335 | 13 | 3.9 | VIL2, EZR | Cytoplasm, cell
membrane, cytoskeleton |
| ARP3 actin-related
protein 3 homolog | 5031573 | 47,797 | 5.61 | 150 | 6 | 3.5 | ACTR3 | Cytoskeleton |
| α-actinin-4 | 2804273 | 102,661 | 5.27 | 72 | 2 | 3.0 | ACTN4 | Cytoplasm,
nucleus |
| Adenylyl cyclase-1
associated protein | 116241280 | 52,222 | 8.27 | 54 | 10 | 2.6 | CAP1 | Cell membrane |
| Fascin | 4507115 | 55,123 | 6.84 | 160 | 19 | 2.5 | FSCN1 | Cytoplasm,
cytoskeleton |
| Cofilin-1 | 5031635 | 18,719 | 8.22 | 129 | 17 | 2.3 | CFL1 | Cytoplasm, nucleus,
cell membrane, cytoskeleton |
| Energy
metabolism |
| Pyruvate Kinase
(Pkm2) | 67464392 | 60,277 | 8.22 | 166 | 20 | 7.3 | PKM2 | Cytoplasm,
nucleus |
| Phosphoglycerate
kinase 1 | 4505763 | 44,985 | 8.30 | 439 | 20 | 6.3 | PGK1, PGKA | Cytoplasm |
| Aldolase A | 28614 | 39,706 | 8.34 | 267 | 11 | 3.5 | ALDOA, ALDA | Cytoplasm,
nucleus |
| α-enolase
(phosphopyruvatehydratase) | 693933 | 47,421 | 7.01 | 148 | 22 | 2.4 | ENO1 | Cytoplasm,
nucleus |
| L-lactate
dehydrogenase A chain | 126047 | 36,950 | 8.44 | 85 | 18 | 2.3 | LDHA, PIG19 | Cytoplasm |
| ATP synthase,
H+ transporting, mitochondrial F1 complex | 4757810 | 59,828 | 9.16 | 182 | 17 | 13.5 | ATP5A1, ATP5F1 | Mitochondrion,
Mitochondrion inner membrane |
|
Dihydrolipoamidesuccinyl transferase | 643589 | 48,896 | 8.90 | 165 | 7 | 3.8 | DLST | Mitochondrion |
| Citrate
synthase | 33337556 | 51,942 | 8.45 | 169 | 5 | 3.4 | CS | Mitochondrion |
| Fumaratehydratase,
mitochondrial | 182794 | 50,524 | 7.23 | 305 | 11 | 2.7 | FH | Mitochondrion,
cytoplasm |
| Glutamate
dehydrogenase | 4885281 | 61,701 | 7.66 | 197 | 16 | 3.9 | GLUD1 | Mitochondrion |
| Dihydrolipoamide
dehydrogenase | 83753870 | 50,656 | 6.50 | 236 | 9 | 2.2 | DLD | Mitochondrion |
| acyl-Coenzyme A
dehydrogenase | 76496475 | 68,414 | 8.76 | 233 | 10 | 2.1 | ACADVL | Mitochondrion,
mitochondrion inner membrane |
| Transketolase | 37267 | 68,435 | 7.90 | 102 | 16 | 2.8 | TKT | Cytoplasm,
nucleus |
| Molecular
chaperone |
| Tumor rejection
antigen 1, Endoplasmin | 74755280 | 92,282 | 4.77 | 89 | 4 | 11.9 | GRP94, TRA1,
HSP90B1 | Endoplasmic
reticulum |
| T-complex protein 1
subunit γ | 14124984 | 60,934 | 6.10 | 157 | 7 | 6.3 | CCT3, CCTG | Cytoplasm |
| Heat shock protein
HSP 90-α | 154146191 | 85,006 | 4.94 | 163 | 5 | 5.2 | HSP90AA1 | Cytoplasm |
| Heat shock protein
HSP 90-β | 119602173 | 57,868 | 4.92 | 59 | 2 | 2.3 | HSP90AB1 | Cytoplasm |
Stress-induced-phosphoprotein)
1 (Hsp70/Hsp90-organizing protein | 5803181 | 63,227 | 6.40 | 145 | 19 | 3.8 | STIP1 | Cytoplasm,
nucleus |
| 60 kDa heat shock
protein | 77702086 | 61,346 | 5.70 | 580 | 16 | 5.0 | HSPD1, HSP60 | Mitochondrion |
| Heat shock
protein | 386785 | 70,110 | 5.42 | 404 | 10 | 4.4 | HSPA1L | Cytoplasm |
| Heat shock 70 kDa
protein 8 | 5729877 | 71,082 | 5.37 | 203 | 13 | 3.8 | HSPA8 | Cytoplasm |
| Stress-70 protein,
mitochondrial | 21264428 | 73,920 | 5.87 | 70 | 6 | 3.7 | HSPA9 | Mitochondrion |
| 78 kDa
glucose-regulated protein | 386758 | 72,185 | 5.03 | 251 | 7 | 4.1 | GRP78, HSPA5 | Endoplasmic
reticulum |
| T-complex
polypeptide 1 | 36796 | 60,869 | 6.03 | 197 | 9 | 4.0 | TCP1 | Cytoplasm,
cytoskeleton |
| Nucleophosmin
(nucleolarphosphoprotein B23, numatrin) | 15214852 | 32,760 | 4.64 | 198 | 12 | 3.3 | NPM1, NPM | Cytoplasm, nucleus,
cytoskeleton |
| Calreticulin | 4757900 | 48,283 | 4.29 | 109 | 12 | 2.3 | CALR | Cytoplasm,
endoplasmic reticulum, extracellular matrix, secreted |
| Structural
molecule |
| Tubulin, β, 2 | 5174735 | 50,255 | 4.79 | 282 | 14 | 3.8 | TUBB2C, TUBB4B | Cytoplasm,
cytoskeleton, microtubule |
| α-tubulin | 37492 | 50,810 | 5.02 | 123 | 13 | 2.6 | TUBA4A, TUBA1 | Cytoplasm,
cytoskeleton, microtubule |
| Cytoskeleton
function |
| Keratin, type I
cytoskeletal 17 | 4557701 | 48,361 | 4.97 | 303 | 28 | 3.7 | KRT17 | Cytoplasm,
intermediate filament, keratin |
| Keratin, type I
cytoskeletal 18 | 30311 | 47,305 | 5.27 | 558 | 28 | 3.5 | KRT18, PIG46
CYK18 | Cytoplasm,
intermediate filament, keratin |
| Keratin, type I
cytoskeletal 19 | 24234699 | 44,079 | 5.04 | 455 | 44 | 2.1 | KRT19 | Intermediate
filament, keratin |
| Keratin, type II
cytoskeletal 6A | 5031839 | 60,293 | 8.09 | 248 | 29 | 5.8 | KRT6A | Intermediate
filament, keratin |
| Keratin, type II
cytoskeletal 2 epidermal | 908801 | 60,448 | 8.09 | 422 | 15 | 4.3 | KRT2 | Intermediate
filament, keratin |
| Keratin, type II
cytoskeletal 8 | 181573 | 53,529 | 5.52 | 238 | 9 | 4.3 | KRT8, CYK8 | Cytoplasm,
Intermediate filament, keratin, nucleus |
| Keratin, type II
cytoskeletal 7 | 12803727 | 51,444 | 5.42 | 287 | 36 | 3.5 | KRT7 | Cytoplasm,
intermediate filament, keratin |
| Transcription
regulation |
| Far upstream
element-binding protein 1 | 17402900 | 67,690 | 7.18 | 111 | 4 | 4.1 | FUBP1 | Nucleus |
| ETS translocation
variant 5 | 221042722 | 65,643 | 5.69 | 174 | 9 | 2.6 | ERM | Nucleus |
| Translation |
| Ribosomal protein
P0 | 4506667 | 34,423 | 5.71 | 193 | 14 | 3.5 | RPLP0 | Cytoplasm,
nucleus |
|
Tyrosyl-tRNAsynthetase | 4507947 | 59,448 | 6.61 | 403 | 16 | 2.4 | YARS | Cytoplasm |
| Heterogeneous
nuclear ribonucleoprotein L | 11527777 | 64,617 | 8.49 | 173 | 6 | 3.9 | HNRNPL, HNRPL,
P/OKcl.14 | Cytoplasm,
nucleus |
| Heterogeneous
nuclear ribonucleoproteins A2/B1 | 4504447 | 36,041 | 8.67 | 188 | 13 | 3.2 | HNRNPA2B1 | Cytoplasm, nucleus,
spliceosome |
| Heterogeneous
nuclear ribonucleoprotein K | 460789 | 51,325 | 5.13 | 88 | 12 | 2.0 | HNRNPK, HNRPK | Cytoplasm, nucleus,
spliceosome |
| Calcium ion binding
protein |
| Annexin A1 | 4502101 | 38,918 | 6.57 | 124 | 26 | 3.9 | ANXA1, ANX1,
LPC1 | Cytoplasm, nucleus,
cell membrane |
| Annexin A2 | 56967118 | 36,634 | 8.32 | 210 | 13 | 2.1 | ANXA2 | Basement membrane,
extracellular matrix |
| Signal
transduction |
| 14-3-3 protein
epsilon | 5803225 | 29,326 | 4.63 | 136 | 26 | 2.5 | YWHAE | Cytoplasm |
| 14-3-3 protein
β/α | 4507949 | 28,179 | 4.76 | 315 | 23 | 2.2 | YWHAB | Cytoplasm |
| Elongation
factor |
| Elongation factor
Tu | 704416 | 49,851 | 7.70 | 229 | 19 | 3.9 | TUFM | Mitochondrion |
| Proteasome
regulatory |
| 26S proteasome
non-ATPase regulatory subunit 12 | 4506221 | 53,270 | 7.53 | 127 | 6 | 3.3 | PSMD12 | Proteasome,
nucleus, cytoplasm |
| GTPase
activation |
| Human rab GDI | 285975 | 51,088 | 5.94 | 347 | 15 | 3.3 | RABGDIB | Cytoplasm |
| Chromatin
regulator |
| Protein
arginine | 20070220 | 73,322 | 5.88 | 43 | 1 | 3.2 | PRMT5 | Cytoplasm,
nucleus |
| N-methyltransferase
5 |
| Glycan
metabolism |
| Protein kinase C
substrate 80K-H isoform 2 | 48255891 | 60,110 | 4.34 | 51 | 8 | 2.6 | PRKCSH | Endoplasmic
reticulum |
| Protein disulfide
isomerase |
| Prolyl
4-hydroxylase, β polypeptide | 20070125 | 57,480 | 4.76 | 586 | 24 | 2.6 | P4HB | Endoplasmic
reticulum |
| Protease
inhibitor |
| Serine proteinase
inhibitor | 62898301 | 42,857 | 5.90 | 167 | 9 | 2.3 | SERPIN | Secreted |
Functional studies of protein expression
in cholangiocarcinoma cells cultured in matrix gel
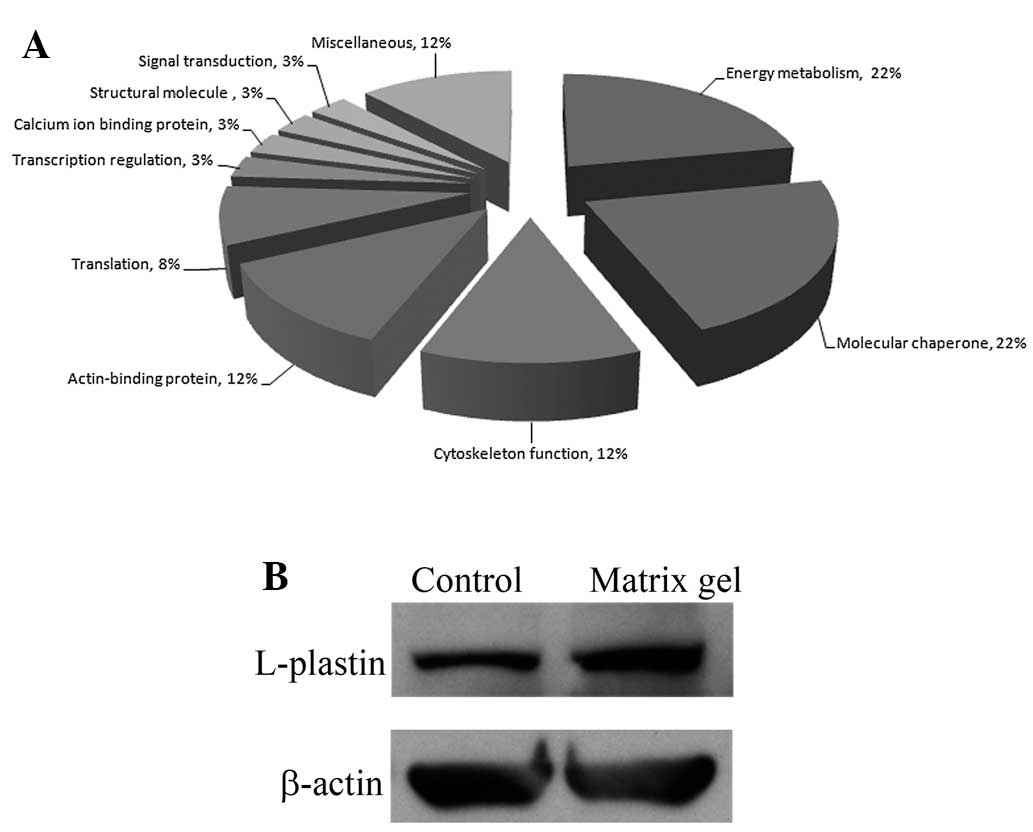
The identified proteins that exhibited significant
changes in expression levels were classified using a UniProtKB
search for protein functional analysis in the species Homo
sapiens (human). Based on the search results, these proteins
are involved in energy metabolism, molecular chaperoning,
cytoskeleton functions, actin binding, translation, transcription
regulation, calcium ion binding, cell structure and signal
transduction (Fig. 2A). Contact
with the ECM and the remodeling of the actin cytoskeleton can drive
cancer cell motility and promote invasion. L-plastin is one of the
actin-binding proteins that exhibited a high level of protein
expression in cholangiocarcinoma cells cultured in matrix gel. We
performed a western blot analysis to confirm the results of the
proteomic study. The results showed that a high level of L-plastin
expression was identified in RMCCA1 cells cultured in matrix gel
(Fig. 2B). A previous study
demonstrated that L-plastin localizes to actin-rich membrane
structures involved in locomotion, adhesion and immune defense,
thereby implying that L-plastin is involved in the organization of
the actin cytoskeleton (17). In
addition, L-plastin has also been detected in solid tumors of
epithelial and mesenchymal origin and has been suggested to be
involved in cancer cell invasion (18). In line with these observations, we
found that the number of cholangiocarcinoma cell invasion events
significantly decreased when the expression of L-plastin was
inhibited with L-plastin siRNA.
Effect of L-plastin on cholangiocarcinoma
cell invasion
To determine whether the expression of L-plastin is
associated with cholangiocarcinoma cell invasion, we knocked down
the expression of L-plastin using L-plastin siRNA. The western blot
(Fig. 3A) and immunofluorescence
studies (Fig. 3B) demonstrated that
L-plastin was significantly downregulated after transfecting the
RMCCA1 cells with L-plastin siRNA. Moreover, the invasion assay
showed that the number of cancer cell invasion events was
significantly decreased with the L-plastin siRNA cells compared
with those treated with the control dsRNA (P<0.001; Fig. 3C).
Detection of L-plastin expression in
paraffin-embedded cholangiocarcinoma specimens
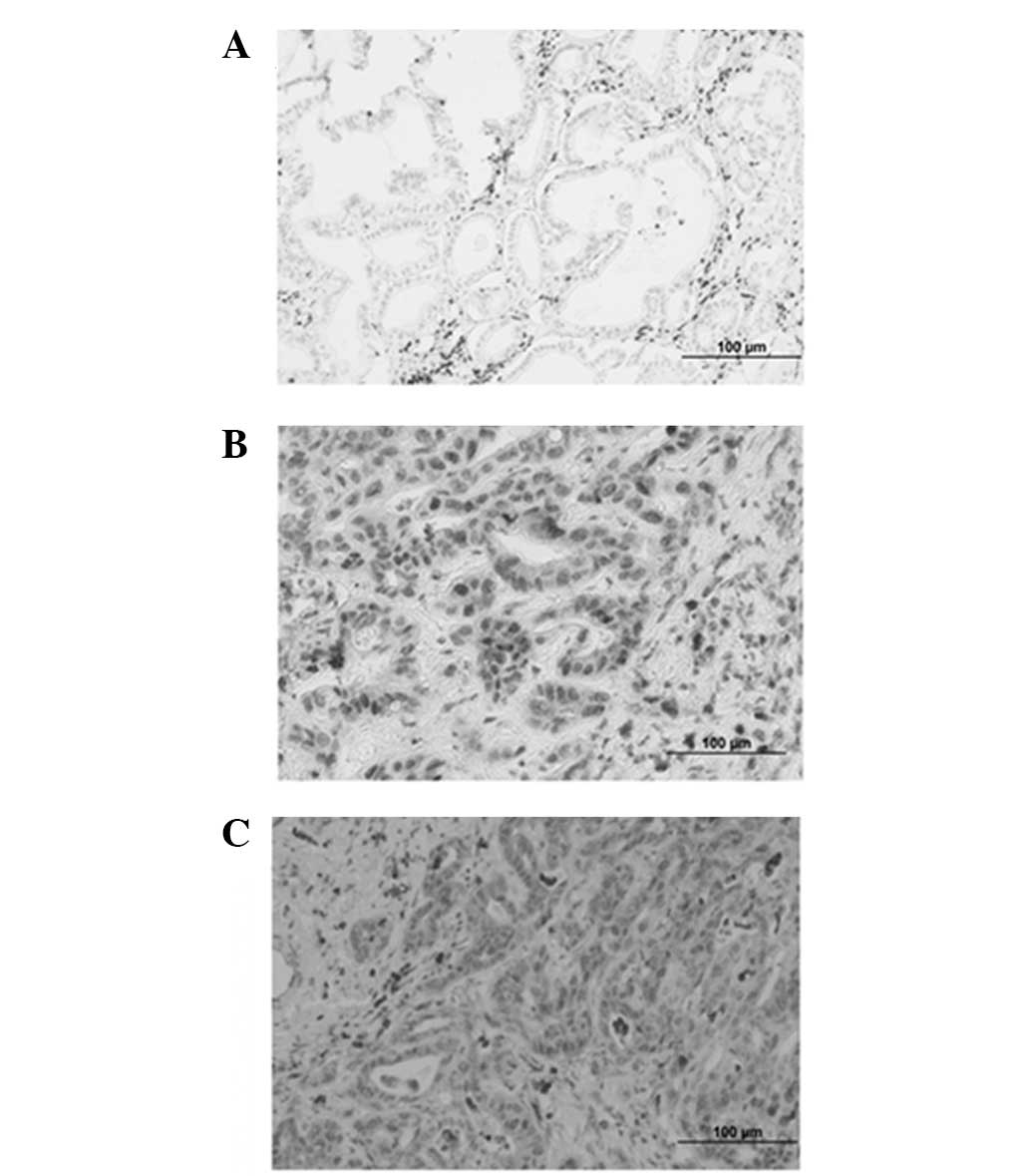
The expression of L-plastin was determined by
immunohistochemistry in 24 paraffin-embedded cholangiocarcinoma
specimens. In these cancerous tissues, L-plastin-specific signals
were localized mainly in the nucleus and cytoplasm of
cholangiocarcinoma cells that invaded the basement membrane and
presented as mesenchymal-like cells (Fig. 4). However, cholangiocarcinoma cells
that were arranged in a granular structure were negative for
L-plastin.
We found that 37.5% (9/24) of the cholangiocarcinoma
specimens were positive for the L-plastin expression signal. The
correlation between the expression of L-plastin and the clinical
characteristics is shown in Table
II. The expression of L-plastin in cholangiocarcinoma was
detected in all stages of the disease.
| Table IICorrelation between L-plastin
expression and the clinicopathological features of
cholangiocarcinoma patients. |
Table II
Correlation between L-plastin
expression and the clinicopathological features of
cholangiocarcinoma patients.
| L-plastin
expression | |
|---|
|
| |
|---|
|
Characteristics | Negative | Positive | P-value |
|---|
| Gender |
| Male | 8 | 5 | 1.00 |
| Female | 7 | 4 | |
| Tumor
differentiation |
| Well | 5 | 3 | 1.00 |
| Moderate and
poor | 10 | 6 | |
| Lymph node
metastasis |
| No | 5 | 4 | 0.65 |
| Yes | 10 | 5 | |
| Distant
metastasis |
| No | 11 | 6 | 1.00 |
| Yes | 4 | 3 | |
Discussion
We report for the first time that the ECM plays a
major role in the regulation of cholangiocarcinoma cell invasion.
Based on 2D electrophoresis results, we identified the proteins
that were upregulated when cholangiocarcinoma cells were cultured
in matrix gel for 24 h. L-plastin, a major F-actin-bundling
protein, was significantly upregulated in matrix-gel-coated plates
compared with uncoated plates. The results were confirmed by
western blotting, as L-plastin exhibited higher expression levels
in RMCCA1 cells cultured in matrix-coated plates. A previous study
demonstrated that L-plastin localizes to actin-rich membrane
structures involved in locomotion, adhesion and immune defense,
thereby implying that L-plastin is involved in the organization of
the actin cytoskeleton (17). In
addition, L-plastin has also been detected in solid tumors of
epithelial and mesenchymal origin and has been suggested to play a
role in cancer cell invasion (18).
In line with these observations, we found that the number of
cholangiocarcinoma cell invasion events significantly decreased
when the expression of L-plastin was inhibited with L-plastin
siRNA.
Confirming the results of prior studies, we observed
that L-plastin is located in the nuclei and cytoplasm of cancer
cells (17,19). The functional relevance of the
nucleocytoplasmic shuttling of L-plastin currently remains unclear.
L-plastin may be involved in the regulation of nuclear actin, which
is an essential component of the pre-initiation complex and
cooperates with polymerases I, II and III in the regulation of gene
expression (20). The formation of
protrusive structures is driven by spatially and temporally
regulated actin polymerization at the leading edge of the cell
(21). Further studies should be
performed to elucidate the involvement of L-plastin localization in
cholangiocarcinoma cells. We found that L-plastin was primarily
expressed in mesenchymal-like cholangiocarcinoma cells. These
findings suggest that L-plastin expression is associated with the
epithelial-mesenchymal transition of cholangiocarcinoma cells.
To understand whether our in vitro findings
are also relevant in vivo, we performed immunohistochemical
analyses of tumor specimens derived from cholangiocarcinoma
patients. Our analyses demonstrated that L-plastin is expressed in
cholangiocarcinoma specimens. However, the level of expression was
not significantly correlated with tumor differentiation, lymph node
or metastatic status. This finding is in contrast to that reported
for colorectal cancer, in which the expression of L-plastin is
significantly correlated with cancer staging (22). Variations in the biological features
of the tumors and the limited number of specimens in our study may
account for these differences. In conclusion, attachment to the ECM
promotes cholangiocarcinoma cell progression by inducing L-plastin
expression. Understanding this mechanism may help to identify a
novel molecular target for the development of an effective therapy
for cholangiocarcinoma patients.
Acknowledgements
The authors would like to thank Ms. Atchara Paemanee
at The National Center for Genetic Engineering and Biotechnology
for the mass spectrometry assistance. This study was supported by
Rajavithi Hospital (Bangkok, Thailand) and the Thailand Graduate
Institute of Science and Technology (TGIST), the National Science
and Technology Development Agency (Pathum Thani, Thailand).
References
|
1
|
Leelawat K, Narong S, Wannaprasert J and
Leelawat S: Serum NGAL to clinically distinguish cholangiocarcinoma
from benign biliary tract diseases. Int J Hepatol.
2011:8735482011.
|
|
2
|
Srisomsap C, Sawangareetrakul P,
Subhasitanont P, et al: Proteomic studies of cholangiocarcinoma and
hepatocellular carcinoma cell secretomes. J Biomed Biotechnol.
2010:4371432010.
|
|
3
|
Sripa B and Pairojkul C:
Cholangiocarcinoma: lessons from Thailand. Curr Opin Gastroenterol.
24:349–356. 2008.
|
|
4
|
Van Beers BE: Diagnosis of
cholangiocarcinoma. HPB (Oxford). 10:87–93. 2008.
|
|
5
|
Kaewpitoon N, Kaewpitoon SJ and Pengsaa P:
Opisthorchiasis in Thailand: review and current status. World J
Gastroenterol. 14:2297–2302. 2008.
|
|
6
|
Chamberlain RS and Blumgart LH: Hilar
cholangiocarcinoma: a review and commentary. Ann Surg Oncol.
7:55–66. 2000.
|
|
7
|
Nakagohri T, Kinoshita T, Konishi M,
Takahashi S and Gotohda N: Surgical outcome and prognostic factors
in intrahepatic cholangiocarcinoma. World J Surg. 32:2675–2680.
2008.
|
|
8
|
Washburn WK, Lewis WD and Jenkins RL:
Aggressive surgical resection for cholangiocarcinoma. Arch Surg.
130:270–276. 1995.
|
|
9
|
Friedl P, Zänker KS and Bröcker EB: Cell
migration strategies in 3-D extracellular matrix: differences in
morphology, cell matrix interactions, and integrin function.
Microsc Res Tech. 43:369–378. 1998.
|
|
10
|
Lomberk G: The extracellular matrix and
cell migration. Pancreatology. 10:4–5. 2010.
|
|
11
|
Painter KJ, Armstrong NJ and Sherratt JA:
The impact of adhesion on cellular invasion processes in cancer and
development. J Theor Biol. 264:1057–1067. 2010.
|
|
12
|
Alvaro D and Mancino MG: New insights on
the molecular and cell biology of human cholangiopathies. Mol
Aspects Med. 29:50–57. 2008.
|
|
13
|
Prakobwong S, Pinlaor S, Yongvanit P,
Sithithaworn P, Pairojkul C and Hiraku Y: Time profiles of the
expression of metalloproteinases, tissue inhibitors of
metalloproteases, cytokines and collagens in hamsters infected with
Opisthorchis viverrini with special reference to peribiliary
fibrosis and liver injury. Int J Parasitol. 39:825–835. 2009.
|
|
14
|
Sripa B, Mairiang E, Thinkhamrop B, et al:
Advanced periductal fibrosis from infection with the carcinogenic
human liver fluke Opisthorchis viverrini correlates with
elevated levels of interleukin-6. Hepatology. 50:1273–1281.
2009.
|
|
15
|
Rattanasinganchan P, Leelawat K,
Treepongkaruna SA, et al: Establishment and characterization of a
cholangiocarcinoma cell line (RMCCA-1) from a Thai patient. World J
Gastroenterol. 12:6500–6506. 2006.
|
|
16
|
Leelawat K, Leelawat S, Narong S and
Hongeng S: Roles of the MEK1/2 and AKT pathways in CXCL12/CXCR4
induced cholangiocarcinoma cell invasion. World J Gastroenterol.
13:1561–1568. 2007.
|
|
17
|
Delanote V, Vandekerckhove J and Gettemans
J: Plastins: versatile modulators of actin organization in
(patho)physiological cellular processes. Acta Pharmacol Sin.
26:769–779. 2005.
|
|
18
|
Samstag Y and Klemke M: Ectopic expression
of L-plastin in human tumor cells: diagnostic and therapeutic
implications. Adv Enzyme Regul. 47:118–126. 2007.
|
|
19
|
Delanote V, Van Impe K, De Corte V, et al:
Molecular basis for dissimilar nuclear trafficking of the
actin-bundling protein isoforms T- and L-plastin. Traffic.
6:335–345. 2005.
|
|
20
|
Ben-Ari Y, Brody Y, Kinor N, et al: The
life of an mRNA in space and time. J Cell Sci. 123:1761–1774.
2010.
|
|
21
|
Yamaguchi H and Condeelis J: Regulation of
the actin cytoskeleton in cancer cell migration and invasion.
Biochim Biophys Acta. 1773:642–652. 2007.
|
|
22
|
Otsuka M, Kato M, Yoshikawa T, et al:
Differential expression of the L-plastin gene in human colorectal
cancer progression and metastasis. Biochem Biophys Res Commun.
289:876–881. 2001.
|