Introduction
Insulinoma is a rare tumor of the alimentary tract
originating from insulin-synthetizing pancreatic beta cells. Its
incidence is estimated at four per million individuals per year
worldwide (1). Although insulinoma
is benign in the majority of cases, it can be malignant in <10%
of patients (2). Typically,
insulinoma manifests as Whipple’s triad, which includes
hypoglycemia, elevated blood levels of insulin accompanied by a
decrease in blood glucose levels to <50 mg/dl, and normalization
of hypoglycemic signs following administration of sugar (3). Due to the low activity or abnormal
structure of synthesized hormones, certain insulinomas remain
asymptomatic until reaching considerable size, when the signs of
their compression on surrounding tissues can be observed (4). Co-existence of insulinoma and diabetes
has rarely been reported (3). This
study presents the case of a female patient with a one month
history of type 2 diabetes, who underwent surgery due to a
pancreatic tumor diagnosed as an insulinoma-type neuroendocrine
pancreatic tumor by histopathological examination. During two years
of follow-up the patient has remained in a good general condition.
The atypical symptomatology and history of the disease, as well as
the associated diagnostic challenges must be emphasized. To the
best of our knowledge, this is only the second case of the
insulinoma with elevated glucose levels in the blood to be reported
in the literature. Patient provided written informed consent.
Case report
Case presentation
A 47-year-old female without a history of acute
pancreatitis and diagnosed with type 2 diabetes one month
previously, was admitted to the Second Department of General and
Gastroenterological Surgery, Medical University of Bialystok
(Bialystok, Poland) due to a tumor of the pancreatic head which had
been diagnosed at the Regional Hospital of Lomza (Lomza, Poland)
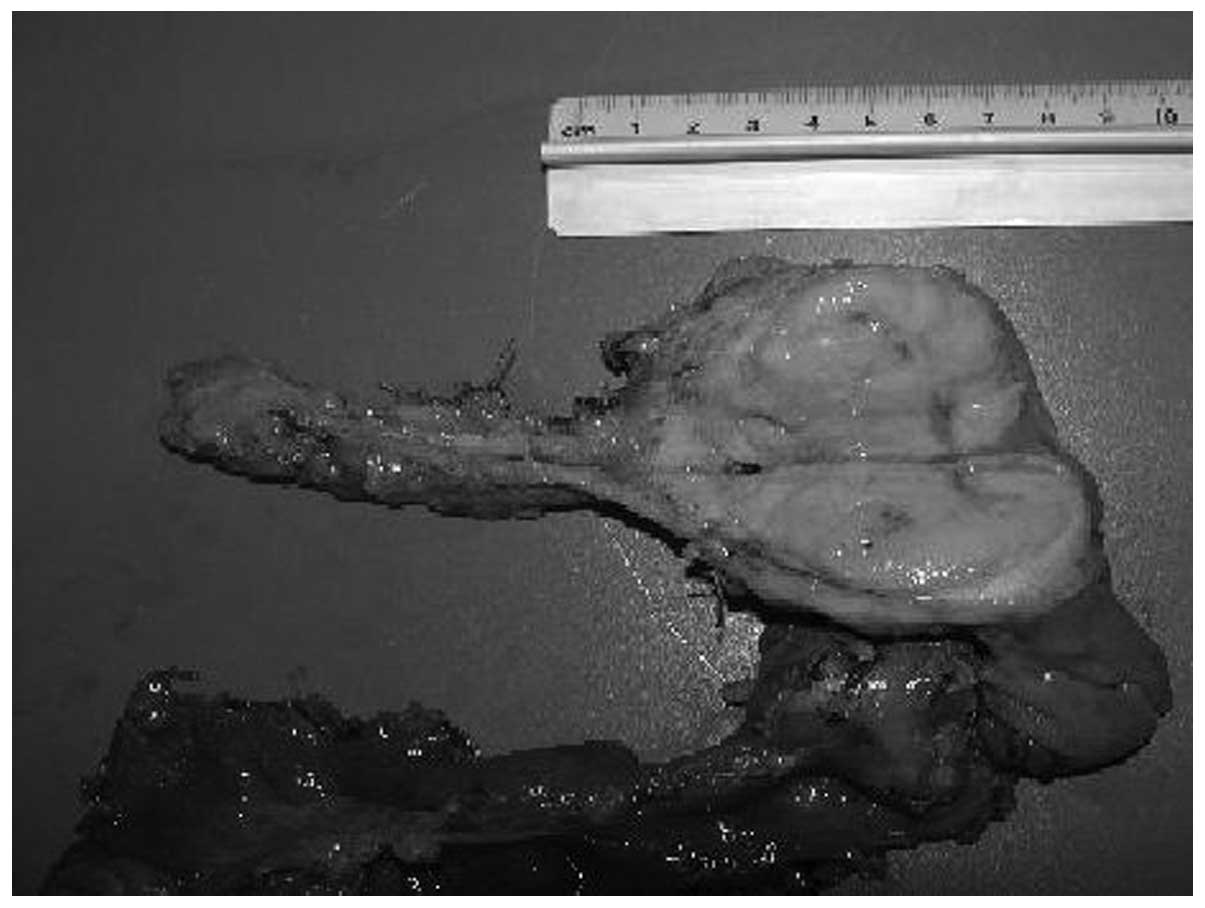
(Fig. 1). On admission, the patient
complained of polydipsia, polyuria and periodical occurrence of
soft stool. Moreover, the patient had lost 3 kg during the past
month. No abnormalities were documented on physical examination.
The patient’s BMI was 21 kg/m2, and laboratory tests
also did not reveal any abnormalities aside from high blood glucose
levels (up to 16.8 mmol/l) and increased C peptide levels (2.17
ng/ml). A 2-h oral glucose tolerance test revealed the same levels
of C peptide (2.14 ng/ml). The concentration of bilirubin was
normal. A 5.5-cm tumor of the pancreatic head was shown on computed
tomography, compressing the interior vena cava and the right renal
vein, and segmentally displacing the duodenal loop. Following
normalization of glycemia with insulin, the patient was qualified
for scheduled surgery.
Surgery
A lard-like, gray-white tumor of the pancreatic
head, measuring 5.5 cm in diameter, was revealed intraoperatively.
The tumor was observed to compress the common bile duct and the
pancreatic duct, and regression of the pancreatic body and tail
parenchyma was evident. No lymph node metastases were documented on
intraoperative microscopic examination. The pancreas was resected
completely. Due to the frozen section examination which revealed a
benign characteristic of the tumor, a pylorus-preserving
pancreticoduodenectomy was performed according to the method of
Traverso and Longmire (5), and the
regional lymph nodes were resected (Fig. 2). Pancreaticoduodenectomy remains
the standard surgical treatment for resectable tumors of the
pancreatic head (5). There was no
postoperative morbidity and, following 12 days of hospitalization,
the patient was discharged from hospital in good overall
status.
Postoperative pathological analysis
Pathomorphological examination of the surgical
specimen revealed a G2 (moderately differentiated) and pT3 (tumor
extends beyond the pancreas, but without involvement of the celiac
axis or superior mesenteric artery) tumor according to the TNM
Staging for Foregut Neuroendocrine Tumors of the Stomach, Duodenum,
and Pancreas (6). The remaining
pancreatic parenchyma showed signs of chronic fibrotic
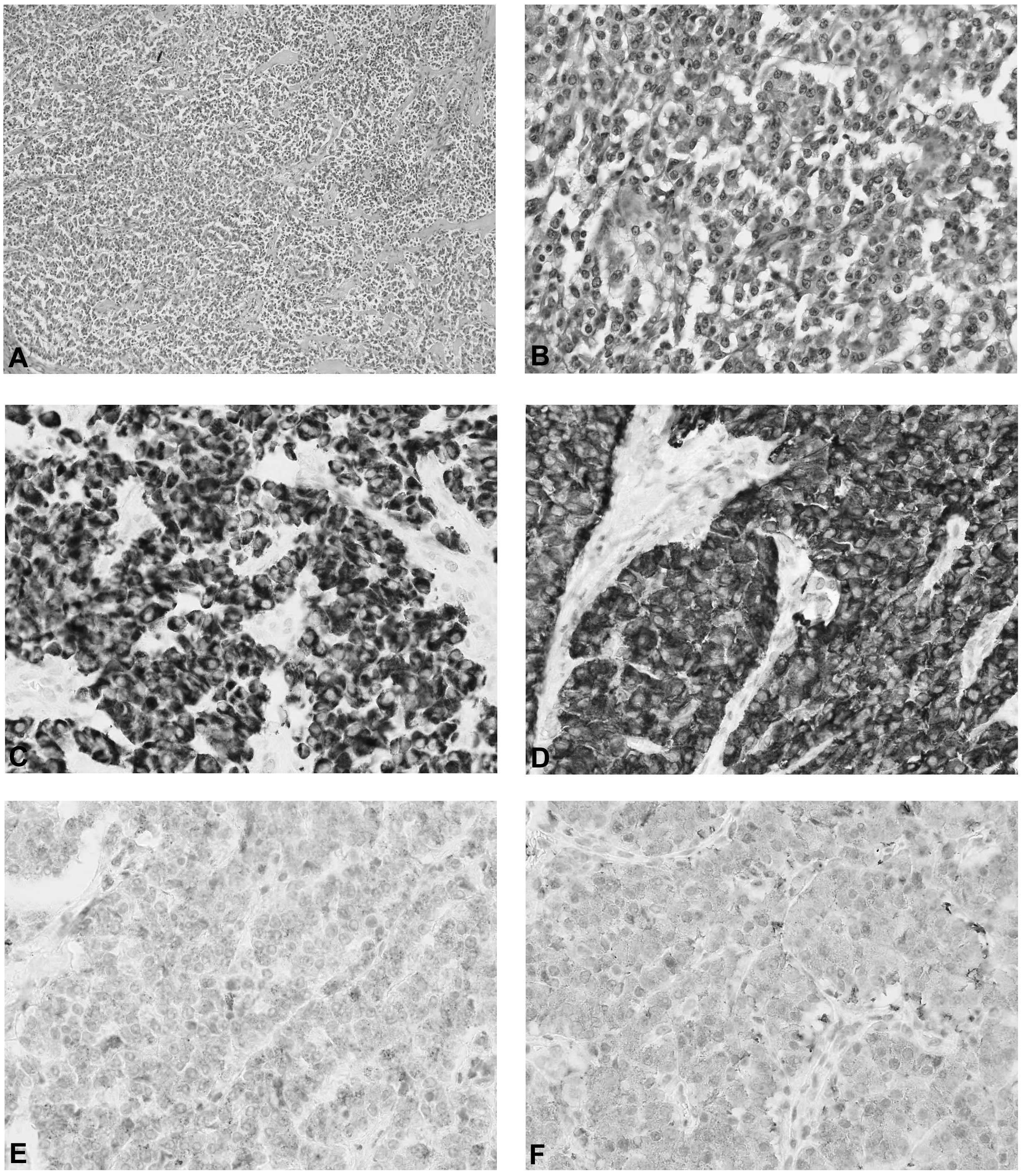
inflammation. Immunohistochemical examination of the tumor revealed
the presence of chromogranin, synaptophysin, neuron-specific
enolase and pancytokeratin (Fig.
3). No metastases were documented in the 16 removed lymph
nodes.
Follow-up
The patient was discharged home in a good general
condition and visited the Surgical Outpatient Clinic for two weeks
following the surgery. The patient was not hospitalized during the
two years of postoperative follow-up. The patient has remains
disease-free without any complaints and with complete control of
diabetes mellitus.
Discussion
Neuroendocrine pancreatic tumors form a group of
heterogenic neoplasms originating from exocrine cells. Although
these tumors more commonly occur spontaneously, they can be
associated with multiple endocrine neoplasia type 1 syndrome in 10%
of cases. Insulinoma is a hormonally active tumor originating from
insulin-synthesizing beta cells of the pancreas. Although it is
typically benign, it can be malignant in 10% of patients. Most
(90%) insulinomas are no larger than 2 cm (7). Due to their small size, the
sensitivity of ultrasound and computed tomography in detection of
insulinoma is low (sensitivity range, 23–63 and 40–73%,
respectively) (8).
Usually, patients are evaluated for potential
insulinoma due to hypoglycemic signs, such as hand tremor,
excessive sweating, heart palpitations, double vision and sudden
loss of consciousness. Moreover, cases in which the initial signs
of insulinoma included seizure episodes or behavioral disorders
have been reported. Abnormalities in laboratory results include
hypoglycemia associated with elevated levels of insulin and high
activity of C peptide, corresponding to the overproduction of
endogenous insulin. The supervised 72-h fast is the gold standard
test for the diagnosis of insulinoma (9). It is necessary to document
hypoglycemia during the test, as insulinoma demonstrates a too high
insulin concentration in the face of hypoglycemia. Patients with
type 2 diabetes in whom the initial manifestation of insulinoma
included a decreased demand for insulin or even a normalization of
glycemia have also been described in literature. However, the
coexistence of insulinoma with hyperglycemia, as in the present
case, has rarely been reported. Both the histopathological
examination of the tumor and elevated C peptide levels were
essential for the diagnosis of insulinoma in the current, non-obese
patient. Normal insulin levels do not exclude the possibility of
the disease, as the absolute insulin levels are not elevated in all
patients with insulinoma (10,11).
Such patients may secrete a variety of insulin precursor and/or its
fragments.
Only one such case was recorded amongst 313
insulinoma-type tumors treated at Mayo Clinic between 1927 and
1993. Moreover, only one case of insulinoma associated with
hyperglycemia was observed among 443 Japanese patients treated for
this tumor between 1976 and 1990 (12). The patient in the present case had
no history of hypoglycemic signs. Neuroendocrine tumors are
frequently asymptomatic. According to the literature, between 0.8
and 10% of tumors are detected during autopsy (13). Frequently, hormonally active tumors
do not present with symptoms specific to a given hormone, due to
the insufficient activity/quantity or the type of hormone (for
example pancreatic polypeptides do not lead to any clinical
symptoms). Kazijan et al (14) identified 50 clinically asymptomatic
cases in a group of 70 patients who underwent surgery for
neuroendocrine pancreatic tumors. Frequently, the initial symptoms
of such tumors include the compression signs associated with their
overgrowth. The tumor detected in the present patient was 5.5 cm in
diameter and compressed the pancreatic duct, the common bile duct
and the duodenum. Additionally, fibrotic pancreatitis was
identified on pathomorphological examination as a potential reason
for the lack of insulin production in destroyed pancreatic islets
and hyperinsulinemia. Atypical clinical manifestation prevented the
establishment of a correct preoperative diagnosis in the present
patient. Sudden onset of diabetes in an otherwise non-obese
patient, rapid weight loss, defecation disorders, and the feeling
of weakness with associated pain could rather suggest pancreatic
adenocarcinoma.
Surgery is the basic treatment option for both
pancreatic adenocarcinoma and neuroendocrine tumors of this organ.
However, the recommendations on the optimal extent of resection
differ. Radical surgery, including resection of the pancreas and
regional lymph nodes, should be performed in adenocarcinoma cases.
By contrast, an enucleation without intact tissue margin is
sufficient in the case of small isolated neuroendocrine tumors,
enabling a 90% five-year survival rate (12). Moreover, lymphadenectomy is not
required in the case of less-advanced neuroendocrine tumors. In the
case of distant metastases, cytoreduction of the endocrine tumor
mass raises the possibility of efficient chemotherapy. In addition,
adenocarcinoma is considered non-resectable and, thus, surgery is
not advised (15). The patient in
the present case presented with clinically asymptomatic, highly
advanced pancreatic insulinoma of considerable size. Such an
unfavorable profile of prognostic factors fully substantiated
radical surgery, which was performed despite the lack of
preoperative diagnosis.
This case confirms that a correct diagnosis can only
be established on the basis of the postoperative pathomorphological
examination. In addition, due to the high malignant potential of
neuroendocrine tumours, radical surgery with regional
lymphadenectomy and intraoperative frozen section evaluation
remains the treatment of choice. In conclusion, the differential
diagnosis of pancreatic neuroendocrine tumours must include
insulinomas with high blood glucose levels, as certain
neuroendocrine tumors are biologically inactive.
References
|
1
|
Service FJ, McMahon MM, O’Brien PC and
Ballard DJ: Functioning insulinoma - incidence recurrence and
long-term survival of patients: a 60-year study. Mayo Clin Proc.
66:711–719. 1991.
|
|
2
|
Abbasakoor NO, Healy ML, O’Shea D, et al:
Metastatic insulinoma in a patient with type 2 diabetes mellitus:
Case report and review of the literature. Int J Endocrinol.
2011:1240782011.
|
|
3
|
Karam MD and Masharani U: Hypoglycemic
disorders. Basic and Clinical Endocrinology. Greenspan FS and
Gardnern DG: 7th edition. McGraw-Hill; New York, NY: pp. 747–766.
2004
|
|
4
|
Chen M, Van Ness M, Guo Y and Gregg J:
Molecular pathology of pancreatic neuroendocrine tumors. J
Gastrointest Oncol. 3:182–188. 2012.
|
|
5
|
Traverso LW and Longmire WP Jr:
Preservation of the pylorus in pancreaticoduodenectomy. Surg
Gynecol Obstet. 146:959–962. 1978.
|
|
6
|
Edge SB, Byrd DR, Compton CC, et al:
Exocrine and Endocrine Pancreas. AJCC Cancer Staging Manual. 7th
edition. Springer; New York, NY: pp. 241–249. 2010
|
|
7
|
Hashimoto LA and Walsh RM: Preoperative
localization of insulinomas is not necessary. J Am Call Surg.
189:369–373. 1999.
|
|
8
|
Ozkaya M, Yuzbasioglu MF, Koruk I, Cakal
E, Sahin M and Cakal B: Preoperative detection of insulinomas: two
case reports. Case J. 1:3622008.
|
|
9
|
Breidalh HD and Rynearson EH: Clinical
aspects of hyperinsulinism. J Am Med Assoc. 160:198–204. 1956.
|
|
10
|
Carneiro DM, Levi JU and Irvin GL III:
Rapid insulin assay for intraoperaive confirmation of complete
resection of insulinomas. Surgery. 132:937–942. 2002.
|
|
11
|
Doherty GM, Doppman JL, Shawker TH, et al:
Results of a prospective strategy to diagnose, localize, and resect
insulinomas. Surgery. 110:989–996. 1991.
|
|
12
|
Ishii H, Ito T, Moriya S, Horie Y and
Tsuchiya M: Insulinoma - a statistical review of 443 cases in
Japan. Nihon Rinsho. 51:199–206. 1993.(In Japanese).
|
|
13
|
Halfdanarson TR, Rabe KG, Rubin J and
Petersen GM: Pancreatic neuroendocrine tumors (PNETs): incidence,
prognosis and recent trend toward improved survival. Ann Onc.
19:1727–1733. 2008.
|
|
14
|
Kazanjian KK, Reber HA and Hines OJ:
Resection of pancreatic neuroendocrine tumors: results of 70 cases.
Arch Surg. 141:765–770. 2006.
|
|
15
|
Kulke MH, Bendell J, Kvols L, Picus J,
Pommier R and Yao J: Evolving diagnostic and treatment strategies
for pancreatic neuroendocrine tumors. J Hematol Oncol.
4:292011.
|