Introduction
Previous studies have shown that cyclooxygenase-2
(COX-2) expression is enhanced in various solid tumors, including
breast, lung and colorectal cancer, and is closely associated with
tumor proliferation, invasion and metastasis, and thus, COX-2 is
considered a promising target in antitumor gene therapy (1,2). As an
important method for investigating gene function, the RNA
interference (RNAi) technique is a rapid, economical and highly
efficient technique for silencing gene expression (3,4). In
the present study, a short hairpin RNA (shRNA) lentiviral vector
was constructed and its effect on malignant biological behaviors,
including proliferation, invasion and metastasis, of breast cancer
cells was investigated. In addition, the function of COX-2 in the
carcinogenesis and development of breast cancer was verified, which
permitted further study regarding these mechanisms.
Materials and methods
Materials
MCF-7 breast cancer cell strains and 293T cells were
purchased from the Shanghai Cell Resource Center of the Chinese
Academy of Sciences (Shanghai, China). The pSPAX2, pMD2G and
pLVX-shRNA1 vectors were purchased from Clontech Laboratories
(Mountain View, CA, USA). The Plasmid Midi kit was purchased from
Qiagen (Valencia, CA, USA). Opi-MEM, Escherichia coli DH5α
and Taq DNA polymerase were purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA). T4 DNA ligase, BamHI and
EcoRI restriction enzymes were purchased from New England
Biolabs (Ipswich, MA, USA). Liposome Lipfectamine 2000, Dulbecco’s
modified Eagle’s medium (DMEM), fetal bovine serum (FBS) and
Trypsin were purchased from Invitrogen Life Technologies. The gel
extraction kit was purchased from Tiangen Biotech (Beijing) Co.,
Ltd. (Beijing, Japan). KOD high fidelity enzyme polymerase chain
reaction (PCR) kit and Taq enzymes were purchased from Toyobo Co.,
Ltd. (Osaka, Japan). DNA ladder was purchased from Fermentas
International, Inc., (Burlington, Canada). The Transwell chamber
was purchased from Chemicon (Temecula, CA, USA).
Cell culture
The MCF-7 breast cancer cells were placed in DMEM
containing l0% FBS, incubated at 37°C in 5% CO2, and
digested and passaged with 0.25% trypsin every 2–3 days. Cells at
the logarithmic phase of growth were used for the experiments.
Design and screening of COX-2 shRNA
The target sequences were designed according to the
COX-2 mRNA sequence obtained from GenBank (http://www.ncbi.nlm.nih.gov/genbank) and the shRNA
design principles. Three pairs of shRNA were designed to target
COX-2 (Table I). The synthesis of
the shRNA was carried out by Hanheng Biological Technology
(Shanghai) Co., Ltd. (Shanghai, China). The shRNA was subsequently
transfected into 293T cells according to the manufacturer’s
instructions for Lipfectamine 2000. The transfection results were
observed under a fluorescence microscope 24 h later. After 36 h,
the cells were collected and the protein was extracted. The most
efficient shRNA was selected according to the results of western
blotting.
 | Table IshRNA sequences specific to COX-2. |
Table I
shRNA sequences specific to COX-2.
| shRNA number | Sequence |
|---|
| COX-2 shRNA-1 |
GCTGAATTTAACACCCTCTAT |
| COX-2 shRNA-2 |
GCAGATGAAATACCAGTCTTT |
| COX-2 shRNA-3 |
CCATTCTCCTTGAAAGGACTT |
Construction and transfection of
COX-2-shRNA lentiviral vector
The most efficient pair of shRNA sequences were
selected as the inference target. A double-stranded DNA fragment,
with cohesive termini of the BamHI and EcoRI
restriction enzymes, and the hairpin sequence of
5′-CCATTCTCCTTGAAAGGACTTTTC AAGAGAAAGTCCTTTCAAGGAGAATGG-3′, was
synthesized in vitro. The fragment was ligated into pGC-LV
vectors and then transfected into E. coli DH5α. Following
amplification and screening, positive clones were sequenced by
Invitrogen Life Technologies, the plasmid was extracted and the
COX-2-shRNA lentiviral vector was recombined. The MCF-7 breast
cancer cells transfected with COX-2-shRNA lentiviral vector were
defined as the knockdown group (COX-2-shRNA), the cells with the
negative control sequences as the mock group and the cells with no
sequence as the blank group.
Total RNA extraction and quantitative PCR
detection
Total RNA was extracted using TRIzol (Invitrogen
Life Technologies) and reverse-transcribed into cDNA. The RNA was
then detected by quantitative PCR. COX-2 and GAPDH primers
(internal control) were synthesized by Hanheng Biological
Technology (Shanghai) Co., Ltd. The following primer sequences were
used: COX-2 forward, 5-CCCTTGGGTGTCAAAGGTAA-3′ and reverse,
5′-GCCCTCGCTTATGATCTGTC-3′; and GAPDH forward,
5′-AGAAAATCTGGCACCACACC-3′ and reverse, 5′-AGAGGCGTACAGGGATAGCA-3′.
The reaction conditions for PCR were as follows: Pre-denaturation
at 95°C for 15 sec, denaturation at 95°C for 5 sec and annealing at
60°C for 30 sec, for 45 cycles. The mixture was then denatured at
95°C for 1 min at the end of the PCR and cooled to 55°C, whereby
the double strands of DNA were able to combine sufficiently.
Between 55 and 95°C the light absorption value was recorded for 4
sec at every 0.5°C, and using these values a melting curve was
generated. Quantitative analysis was performed using the ratio of
the target gene to GAPDH. The data were analyzed using the
2−ΔΔCt method.
Analysis of protein expression by western
blotting
Total protein was isolated 72 h after transfection.
Protein quantification was performed using the bicinchoninic acid
assay. The protein sample was normalized simultaneously. The sample
load was 30 μg total protein per lane. Protein from 10% SDS-PAGE
gel was transferred to a polyvinylidene difluoride membrane
following electrophoresis. The protein was blocked with 5% skimmed
dried milk at 4°C. Next, the primary rabbit monoclonal anti-COX-2
(1:500), anti-vascular endothelial growth factor (VEGF)-A (1:800),
anti-VEGF-C (1:800) and anti-GAPDH (1:4,000) antibodies (Cell
Signaling Technology, Inc., Danvers, MA, USA) were added and the
mixture was incubated overnight at 4°C on a rocking platform.
Subsequent to being washed, the membrane was added together with
the horseradish peroxidase-conjugated secondary antibody (1:4,000)
and incubated for 2 h. The membrane was then developed using an
enhanced chemiluminescence system (Pierce Biotechnology, Inc.,
Rockford, IL, USA) and exposed to X-ray film. The gray scales were
then scanned using ImageJ software (National Institutes of Health,
Bethesda, MD, USA).
Determination of cell proliferation by
MTT assay
The MCF-7 breast cancer cells at the logarithmic
phase of growth from each group were seeded into 96-well plates at
100 μl/well, at a density of 1×104 cells/well. The
plates were incubated at 37°C in an atmosphere of 5%
CO2, with saturated humidity, and an MTT assay was
performed 2 to 72 h after incubation. Optical density (OD) values
were detected at a wavelength of 570 nm using a microplate
spectrophotometer (UV1700; Shimadzu Corporation, Kyoto, Japan). The
mean value of five wells was the final OD value used. The cell
proliferation curve was generated with time as the horizontal axis
and OD value as the vertical axis. The suppression rate of
proliferation of the breast cancer cells was calculated as follows:
Suppression rate (%) = [(1 - OD value of the COX-2 - shRNA group) /
OD value of blank group] × 100.
Detection of the invasive capacity of
breast cancer cells by cell invasion assay
Matrigel artificial substrate was layered in the
Transwell chamber. Cell suspension (200 μl) containing
1×105 MCF-7 breast cancer cells from each group was
added to the upper chamber and 10% FBS DMEM medium was added to the
lower chamber. The chamber was incubated at 37°C, in an atmosphere
of 5% CO2 for 48 h to fix and stain the cells. Images
were captured with an optical microscope (magnification, ×100). The
numbers of the cells in the center and the surrounding five zones
were counted and the average was identified as the number of cells
penetrating through the plasma membrane. Each experiment was
performed on three wells and the same experiment was performed in
triplicate.
Detection of the cell migratory capacity
by scratch test
Horizontal lines were scratched across the wells at
the back of the 96-well plate using a marker pen and at least two
lines were scratched for each well. A total of ~5×104
cells were added to each well. The next day, lines perpendicular to
the horizontal lines were scratched with the head of a pipette. The
cells were washed twice with phosphate-buffered saline, and the
sloughing cells were removed. Next, serum-free medium was added
into the wells and the cells were incubated for 24 h. Images were
captured under a fluorescence microscope. The vertical distance of
the inner face of the scratch zone was measured. The cell migrated
number was calculated as follows: Cell migrated number (%) =
(vertical distance of the inner face of the scratch zone prior to
repair - vertical distance of the inner face of the scratch zone
following repair) / vertical distance of the inner face of the
scratch zone prior to repair × 100.
Statistical analysis
Data were analyzed using SPSS version 16.0 (SPSS,
Inc., Chicago, IL, USA). Quantitative data are expressed as the
mean ± standard deviation. The differences between groups were
analyzed by variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
Screening of the COX-2-shRNA lentiviral
vector
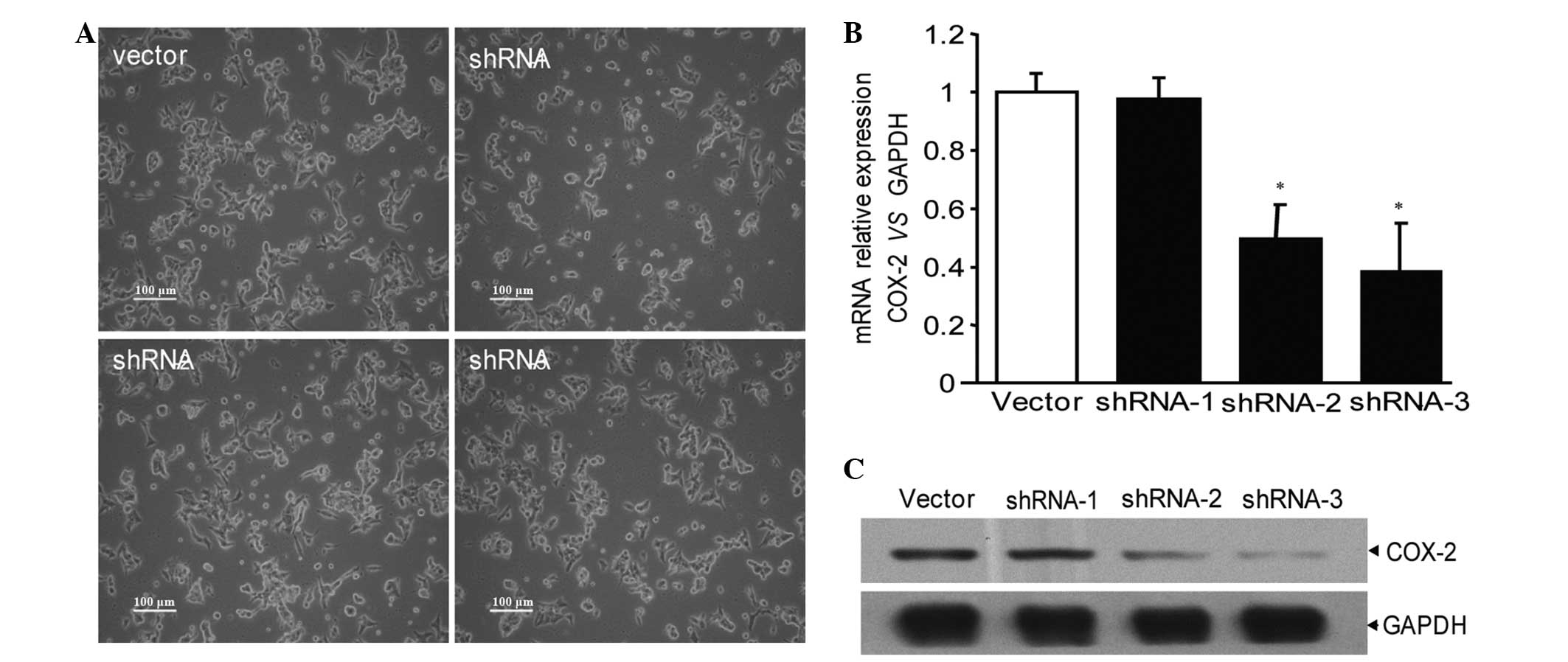
Vectors carrying plasmids with no sequence and
COX-2-shRNA-1, 2 and 3 with three pairs of COX-2 shRNA, were
effectively transfected into the MCF-7 breast cancer cells and
emitted green fluorescence (Fig.
1A). According to western blotting and quantitative PCR
detection, shRNA-1 exhibited no significant interference, shRNA-2
demonstrated partial interference and shRNA-3 exhibited the most
significant interference (Fig. 1B and
C). Therefore, the shRNA-3 sequence was used to recombine shRNA
plasmid vectors. Next, the COX-2-shRNA plasmid was transfected into
the MCF-7 breast cancer cells, then screened and amplified via G418
for further study.
Effects of COX-2-shRNA on COX-2 mRNA in
MCF-7 breast cancer cells
As shown by quantitative PCR, the expression of
COX-2 mRNA in the MCF-7 breast cancer cells of the COX-2-shRNA
group was significantly lower than that of the blank and mock
groups (P<0.05), with no significant difference identified
between the mock and blank groups (Fig.
2A). Western blotting indicated that the expression of the
COX-2 protein in the MCF-7 breast cancer cells of the COX-2-shRNA
group was also significantly lower than that of the blank and mock
groups (P<0.05), which was consistent with the results of the
quantitative PCR (Fig. 2B).
Effects of COX-2-shRNA on the
proliferation of MCF-7 breast cancer cells
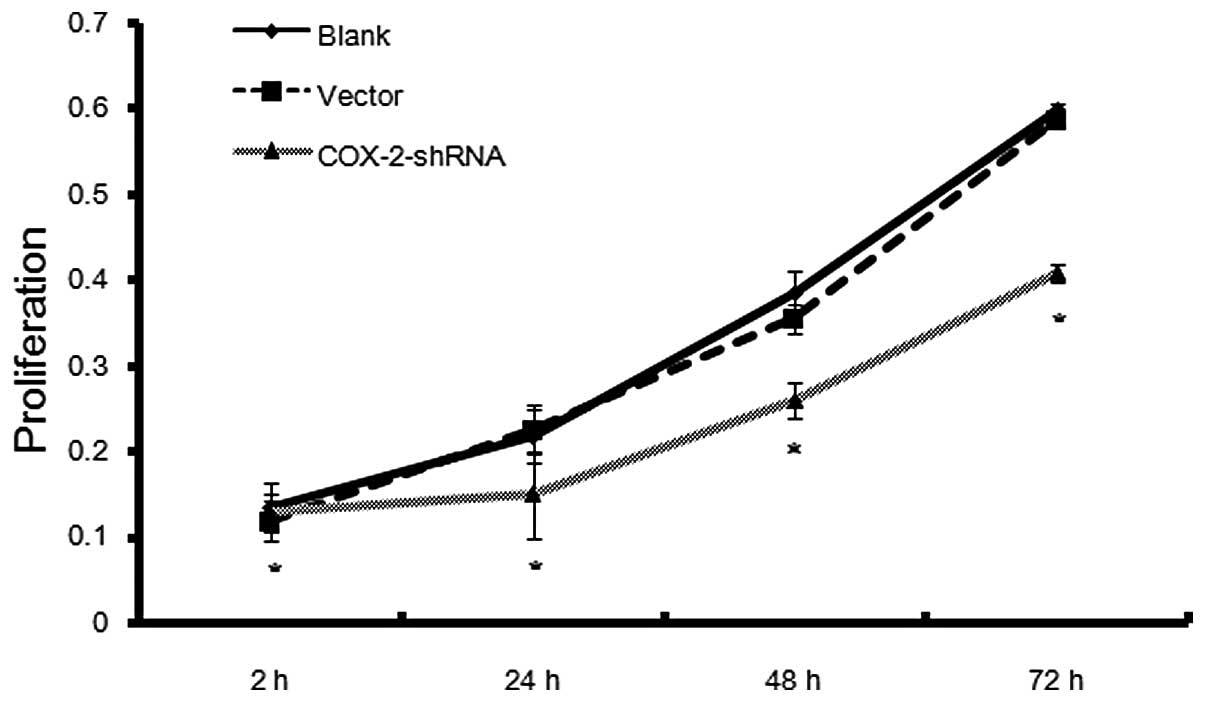
A cell proliferation curve was generated based on
the absorbance values of the MCF-7 breast cancer cells of the
COX-2-shRNA, mock and blank groups, which were measured over 72 h.
The initial absorbance values of the COX-2-shRNA, blank and mock
groups were 0.0986±0.0076, 0.0994±0.0186 and 0.1037±0.0134,
respectively, and no significant differences were identified
(P>0.05). The absorbance values on day three for the
COX-2-shRNA, blank and mock groups were 0.4949±0.0308,
0.6628±0.0245 and 0.6545±0.0155, respectively. No significant
difference in cell proliferation rate was identified between the
blank and mock groups (P>0.05), however, the cell proliferation
rates of the COX-2-shRNA group were significantly lower than that
of the other two groups (P<0.05). The suppression rates 24, 48
and 72 h after COX-2 interference were 24.47, 22.19 and 25.34%,
respectively (Fig. 3).
Changes in the invasive capacity of
breast cancer cells following COX-2 interference
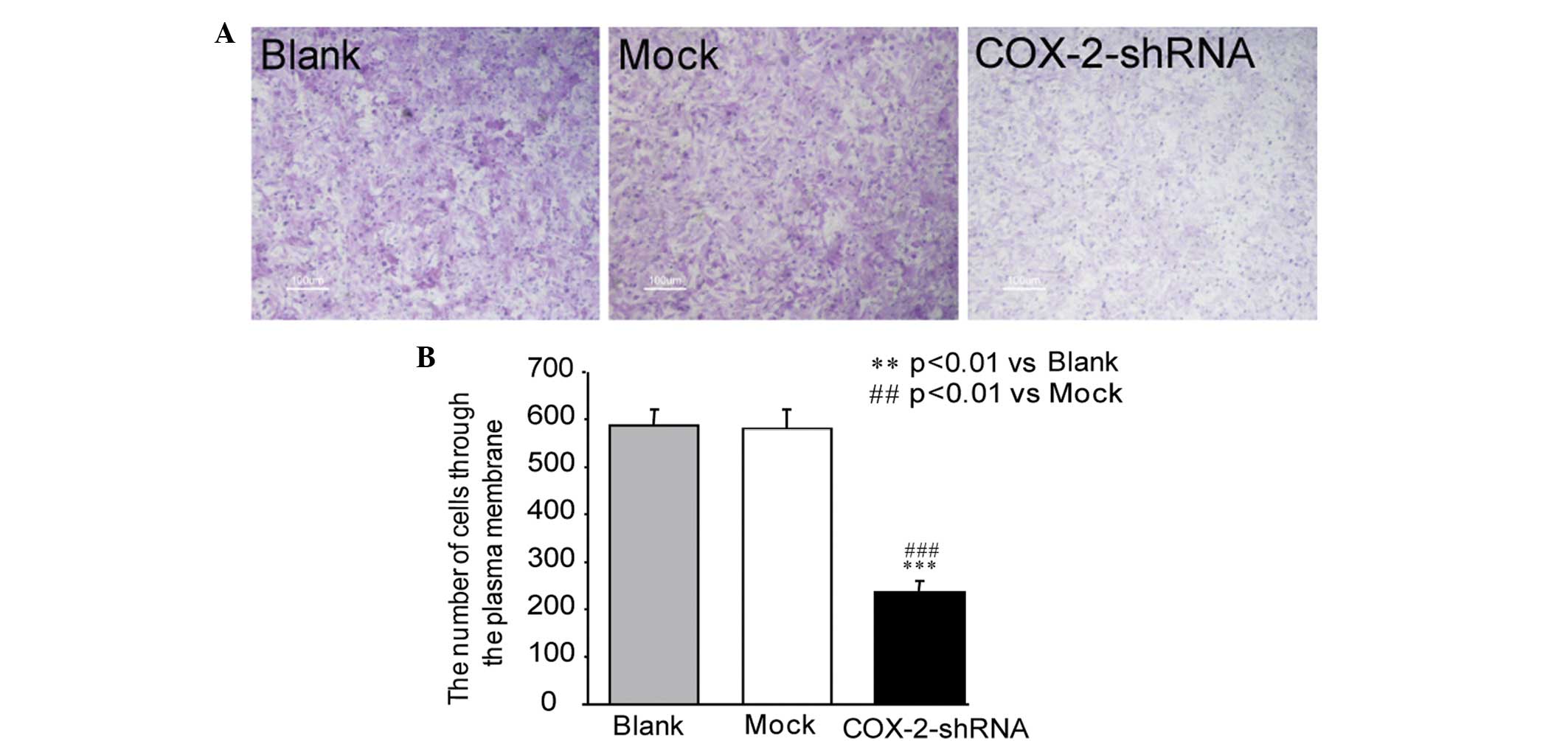
The Transwell assay demonstrated that the number of
MCF-7 breast cancer cells penetrating through the plasma membrane
in the COX-2-shRNA group was 235.5±25.6 at 24 h post-transfection,
which was significantly lower than that of the blank (587.3±35.2)
and mock (580.5±40.7) groups (P<0.05) (Fig. 4A). The results revealed that the
invasive capacity of the MCF-7 breast cancer cells significantly
decreased following transfection with COX-2-shRNA (Fig. 4B).
Changes in the migratory capacity of
breast cancer cells following COX-2 interference
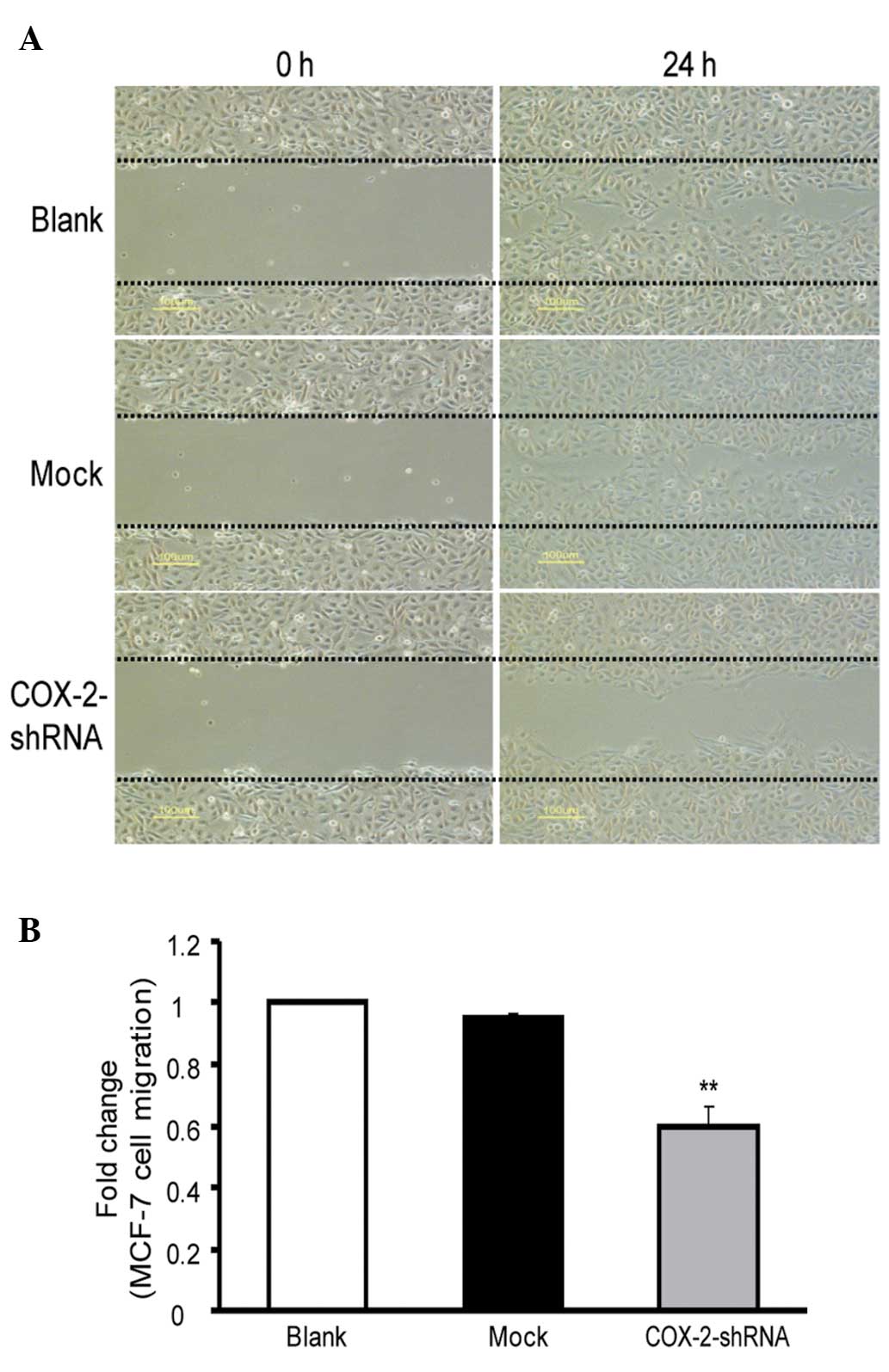
The cell migration of the MCF-7 breast cancer cells
in the COX-2-shRNA group was markedly decreased (Fig. 5A). The cell migration at 24 h in the
COX-2-shRNA group (59.6±7.0%) was significantly lower than that of
the blank (100±0%) and mock (94.7±2.1%) groups, and the differences
were statistically significant (P<0.05). However, no significant
differences were identified between the blank and mock groups
(P>0.05; Fig. 5B). These results
revealed that the cell migration capacity of the MCF-7 breast
cancer cells decreased significantly following transfection with
COX-2-shRNA.
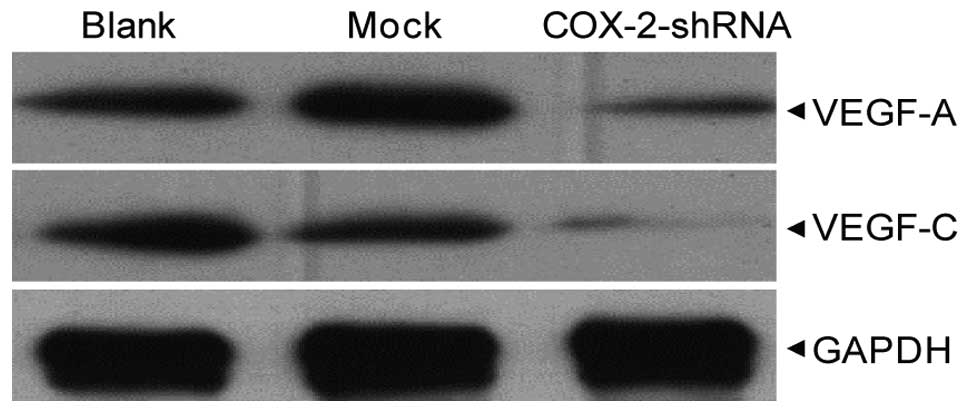
VEGF-A and -C expression following COX-2
interference
Western blotting revealed that following COX-2-shRNA
transfection, the expression of the VEGF-A and VEGF-C proteins in
the breast cancer MCF-7 cells of the COX-2-shRNA group was
significantly lower than that of the blank and mock groups
(P<0.05), however, no significant differences were identified
between the blank and mock groups (P>0.05) (Fig. 6).
Discussion
Breast cancer has become a serious threat to the
health of females worldwide (5). In
China, the incidence of breast cancer is increasing at an annual
rate of 3%, far surpassing lung cancer to become the malignant
tumor with the fastest growing female mortality rate (6). The widely used comprehensive
treatments of surgery, internal medical treatment and radiation
therapy have yielded positive results, however, their efficacy is
beginning to plateau (7). Studies
of molecular tumor biology have identified that breast cancer is a
genetic disease. The activation of oncogenes and the inactivation
of tumor suppressor genes in the regulation of the cellular
physiological processes leads not only to abnormal cell
proliferation and differentiation, but also to defective apoptosis
and drug resistance (8,9). Therefore, it is of great importance
for the prevention and treatment of breast cancer to identify novel
targets for breast cancer gene therapy (5,10).
Previous studies have shown that COX-2 is abnormally expressed in
various tumors, and is directly or indirectly involved in
carcinogenesis and the development of tumors. The association of
COX-2 with breast cancer has been an intense focus of previous
studies (11,12).
COX-2 is an important rate-limiting enzyme in
prostaglandin (PG) synthesis. At least two types of isoenzymes
(COX-1 and COX-2) have been identified in mammals. COX-1 is a
structural gene, which is expressed in normal tissue and cells and
is involved in normal physiological functions. COX-2 is an
inducible enzyme, which is undetectable in the majority of tissues
under normal physiological conditions and which is only rapidly
produced in specific cells when stimulated by mitogens, including
cytokines, endotoxin, carcinogens and oncogenes (13,14).
Previous studies have found that the carcinogenesis mechanism of
COX-2 in breast cancer is complex and that COX-2 exhibits an
important biological role in the proliferation, invasion and
metastasis of breast cancer cells, as well as the regulation of the
activity of relevant factors. Studies by Howe et al
(15) and Singh and Lucci (16) have shown that COX-2 regulates PG
synthesis, that its overexpression increases PG production,
stimulating cell proliferation and promoting tumor formation, and
that PGE2 and PGF2α, among others, stimulate Balb/C3T3 fibroblast
mitosis together with epidermal growth factor (EGF). In addition,
in the presence of EGF, PGE1 and PGE2 stimulate the growth of
breast cells, and PGE2 functions as an epithelial cytokinin,
directly stimulating the proliferation and growth of breast cells
by increasing the levels of estrogen. Gabbert et al
(17) revealed that tumor cells,
when proliferating due to stimulation, increase the number of tumor
cells with invasive potential and promote the division of cells
surrounding the tumor margin, which creates the opportunity for
tumor cell dissociation, and thus enhances the proliferation of
infiltrating cells that form expansive tumor cell nests, to
complete the process of invasion and metastasis. Takahashi et
al (18) demonstrated that
COX-2 enhances tumor proliferation and growth, as well as invasion
and metastasis. Further studies by Sivula et al (19) and Bailey et al (20) have shown that by regulating the
migration of tumor cells, enhancing the degradation of
extracellular matrix and other activities, COX-2 increased the
invasive capacity of cancer cells, further promoting the invasion
of blood and lymphatic vessels, enhancing the transfer of cells to
the lymph nodes and distant organs. The enhanced invasiveness is
associated with the activity of tumor cell matrix metalloproteinase
(MMP)-2, increased expression of MMP-1 mRNA, changes in urokinase
plasminogen activator expression, cell adhesion molecule E-cadherin
deficiency and the increased expression of hyaluronic acid
receptor, CD44, on the cellular surface of tumor invasion and
metastasis molecules. Takahashi et al (18) revealed that the breast cancer cell
strain, Hs578T, with stable COX-2 expression exhibits an increase
in the activity of MMP, thus increasing the capacity to digest the
basement membrane, which provides more direct evidence for the
involvement of COX-2 in cancer cell invasion. These results were
consistent with the study by Hiraga et al (21). Tumor proliferation and growth,
resistance to apoptosis and invasion and metastasis are closely
associated with the formation of tumor blood and lymphatic vessels.
VEGF and COX-2 exhibit multi-channel connections at the gene and
protein expression levels. As demonstrated by Musto (22), colocalizations are frequently
identified between the VEGF-3 and COX-2 genes, which indicates that
a mechanism exists within the tumor cells that controls the
expression of the two genes. Pai et al (23) used molecular biology to demonstrate
that VEGF induces the expression of COX-2, and stabilizes COX-2
mRNA and protein expression via the COX-2 promoter, GATA-related
locus, in vascular endothelial cells. In addition, it is
hypothesized that the VEGF-induced increase in COX-2 occurs via the
activation of p38 mitogen-activated protein kinase and c-Jun
N-terminal kinase factor signaling. It is generally accepted that
COX-2 is involved in the formation of tumor blood vessels. COX-2
significantly promotes the generation of angiogenic factors,
including VEGF, basic fibroblast growth factor, transforming growth
factor-1, platelet-derived growth factor and endothelin-1.
Furthermore, Liu et al (24)
and Uefuji et al (25)
demonstrated that COX-2 increases VEGF-A expression in tumors and
that COX-2 inhibition suppresses VEGF-A expression. In addition,
Bamba et al (26) revealed
that COX-2 acts on the associated receptors by promoting the
synthesis of PGs, including PEG2 and 15-deoxy-PG J2, or induces the
increase of VEGF-A-based angiogenesis factor expression by entering
the nucleus directly via nuclear receptors to induce the formation
of tumor blood vessels. As a member of the VEGF family, VEGF-C was
the first lymphangiogenesis factor to be identified. It induces
proliferation and migration of lymphatic endothelial cells and
promotes lymphatic extension and nascent lymphatic sinus growth via
the MEK/ERK and PI32 kinase/Akt pathways following binding to the
VEGFR-3 receptor. Furthermore, in tumor lymphangiogenesis, VEGF-C
induces internal and surrounding lymphangiogenesis, and promotes
the growth and metastasis of lymphatic tumors (27). Su et al (28) revealed that COX-2 and VEGF-C
expression in a human tumor cell line showed that VEGF-C was
significantly higher in cell lines that overexpress COX-2, and
further studies showed that COX-2 may increase VEGF-C expression
via EP1 and human epidermal growth factor receptor 2 to promote
tumor lymphangiogenesis and lymph node metastasis. Therefore,
inhibition of the COX-2 gene may inhibit the generation of tumor
blood and lymphatic vessels and suppress tumor invasion and
metastasis, as well as proliferation and growth.
RNAi is the most effective antisense technique at
present, originating from a hereditary phenomenon widely existing
in flora and fauna, and serving as a protective mechanism against
the gene instability caused by viral infection and insertion
mutations. The technique specifically induces the degradation of
target mRNA using double-stranded siRNA. In comparison to other
gene knockout techniques, RNAi exhibits high efficiency, stability,
specificity, hereditability and transmissibility, and therefore is
significant in the research of gene function and in tumor gene
therapy (29,30). In the present study, quantitative
PCR and western blotting demonstrated that COX-2-shRNA effectively
suppressed the expression of COX-2 mRNA and protein in MCF-7 breast
cancer cells. In addition, the MTT assay revealed that the
COX-2-shRNA sequence altered the proliferation and growth of the
cells. In particular, the suppression rate of the MCF-7 breast
cancer cells was 24.47, 22.19 and 25.34%, 24, 48, and 72 h after
COX-2 interference, respectively. The present study demonstrated
the importance of COX-2 in maintaining and promoting the
proliferation and growth of breast cancer cells at the mRNA and
protein levels. Following the transfection of COX-2-shRNA into the
breast cancer cells, the invasion and migration capacities were
significantly altered, as shown by the markedly decreased cell
membrane-penetrating capacity and erasion trace repair rates. All
data demonstrated the importance of COX-2 in the invasion and
migration of breast cancer cells. In addition, as shown in the
literature, in the COX-2-shRNA group, the mRNA and protein
expression was reduced significantly and the protein expression of
VEGF-A and VEGF-C was also markedly decreased when compared with
that of the other groups. Therefore, COX-2 downregulation via RNAi
is one of the predominant mechanisms that inhibit the malignant
biological behaviors of breast cancer by reducing the activity of
VEGF-A and VEGF-C, which promote tumor angiogenesis and
lymphangiogenesis. Numerous studies have demonstrated that COX-2
inhibitors exhibit a positive effect against breast cancer.
McCormick et al (31) found
that indomethacin can reduce the incidence of breast cancer and
tumors induced by dimethyl-benzanthracene (DMBA). Harris et
al (32) used celecoxib in a
DMBA-induced breast cancer model and found that the drug markedly
delayed the occurrence of tumors, and that this was more effective
when compared with ketoprofen. Furthermore, Nakatsugi et al
(33) demonstrated that nimesulide,
another COX-2 inhibitor, decreases the incidence of breast cancer
by 28% in a rat model. Harris et al (34) revealed that the administration of
non-steroidal anti-inflammatory drugs (NSAIDs) for between five and
nine years reduces the incidence of breast cancer by 21%, and that
administration for >10 years reduces the incidence by 28%. In
addition, Khuder and Mutgi (35)
recorded that NSAIDs reduced the risk of breast cancer, with a
coefficient of relative risk factor of 0.8 (95% confidence
interval, 0.75–0.89).
In conclusion, the downregulation of COX-2 gene
expression suppresses the malignant biological behavior of breast
cancer cells. Further studies investigating the association between
COX-2 and breast cancer may identify methods of regulating COX-2
expression to prevent and control breast cancer, thus presenting
novel approaches for breast cancer prevention and treatment
(36).
Acknowledgements
This study was supported by grants from the Youth
Science and Research Project of the Health Department of Fujian
Province (grant no. 2009-2-23).
References
|
1
|
Shehzad A, Lee J and Lee YS: Curcumin in
various cancers. Biofactors. 39:56–68. 2013.
|
|
2
|
Dhakal HP, Naume B, Synnestvedt M, et al:
Expression of cyclooxygenase-2 in invasive breast carcinomas and
its prognostic impact. Histol Histopathol. 27:1315–1325. 2012.
|
|
3
|
Ashihara E: RNA interference for cancer
therapies. Gan To Kagaku Ryoho. 37:2033–2041. 2010.(In
Japanese).
|
|
4
|
Liu JL, Wei W, Tang W, et al: Silencing of
lysyl oxidase gene expression by RNA interference suppresses
metastasis of breast cancer. Asian Pac J Cancer Prev. 13:3507–3511.
2012.
|
|
5
|
Engebraaten O, Moen Vollan HK and
Børresen-Dale AL: Triple-negative breast cancer and the need for
new therapeutic targets. Am J Pathol. 183:1064–1074. 2013.
|
|
6
|
Li N, Zheng RS, Zhang SW, et al: Analysis
and prediction of breast cancer incidence trend in China. Zhonghua
Yu Fang Yi Xue Za Zhi. 46:703–707. 2012.(In Chinese).
|
|
7
|
Sheri A and Johnston S: New developments
and future directions in systemic therapy. Clin Oncol (R Coll
Radiol). 25:117–126. 2013.
|
|
8
|
Veeck J1, Noetzel E, Bektas N, et al:
Promoter hypermethylation of the SFRP2 gene is a high-frequent
alteration and tumor-specific epigenetic marker in human breast
cancer. Mol Cancer. 7:832008.
|
|
9
|
Zhang X and Munster PN: New protein kinase
inhibitors in breast cancer: afatinib and neratinib. Expert Opin
Pharmacother. 15:1277–1288. 2014.
|
|
10
|
De Los Santos JF, Cantor A, Amos KD, et
al: Magnetic resonance imaging as a predictor of pathologic
response in patients treated with neoadjuvant systemic treatment
for operable breast cancer. Translational Breast Cancer Research
Consortium trial 017. Cancer. 119:1776–1783. 2013.
|
|
11
|
Taromaru GC, DE Oliveira VM, Silva MA, et
al: Interaction between cyclooxygenase-2 and insulin-like growth
factor in breast cancer: A new field for prevention and treatment.
Oncol Lett. 3:682–688. 2012.
|
|
12
|
Park BW, Park S, Park HS, et al:
Cyclooxygenase-2 expression in proliferative Ki-67-positive breast
cancers is associated with poor outcomes. Breast Cancer Res Treat.
133:741–751. 2012.
|
|
13
|
Huang ZF, Massey JB and Via DP:
Differential regulation of cyclooxygenase-2 (COX-2) mRNA stability
by interleukin-1 beta (IL-1 beta) and tumor necrosis factor-alpha
(TNF-alpha) in human in vitro differentiated macrophages. Biochem
Pharmacol. 59:187–194. 2000.
|
|
14
|
Howe LR, Subbaramaiah K, Brown AM and
Dannenberg AJ: Cyclooxygenase-2: a target for the prevention and
treatment of breast cancer. Endocr Relat Cancer. 8:97–114.
2001.
|
|
15
|
Singh B and Lucci A: Role of
cyclooxygenase-2 in breast cancer. J Surg Res. 108:173–179.
2002.
|
|
16
|
Oliveira VM, Piato S and Silva MA:
Correlation of cyclooxygenase-2 and aromatase immunohistochemical
expression in invasive ductal carcinoma, ductal carcinoma in situ,
and adjacent normal epithelium. Breast Cancer Res Treat.
95:235–241. 2006.
|
|
17
|
Gabbert H, Wagner R, Moll R and Gerharz
CD: Tumor dedifferentiation: an important step in tumor invasion.
Clin Exp Metastasis. 3:257–279. 1985.
|
|
18
|
Takahashi Y, Kawahara F, Noguchi M, et al:
Activation of matrix metalloproteinase-2 in human breast cancer
cells overexpressing cyclooxygenase-1 or -2. FEBS Lett.
460:145–148. 1999.
|
|
19
|
Sivula A, Talvensaari-Mattila A, Lundin J,
et al: Association of cyclooxygenase-2 and matrix
metalloproteinase-2 expression in human breast cancer. Breast
Cancer Res Treat. 89:215–220. 2005.
|
|
20
|
Bailey T, Biddlestone L, Shepherd N, et
al: Altered cadherin and catenin complexes in the Barrett’s
esophagus-dysplasia-adenocarcinoma sequence: correlation with
disease progression and dedifferentiation. Am J Pathol.
152:135–144. 1998.
|
|
21
|
Hiraga T, Myoui A, Choi ME, et al:
Stimulation of cyclooxygenase-2 expression by bone-derived
transforming growth factor-beta enhances bone metastases in breast
cancer. Cancer Res. 66:2067–2073. 2006.
|
|
22
|
Musto P: Thalidomide therapy for
myelodysplastic syndromes: current status and future perspectives.
Leuk Res. 28:325–332. 2004.
|
|
23
|
Pai R, Szabo IL, Soreghan BA, et al:
PGE(2) stimulates VEGF expression in endothelial cells via
ERK2/JNK1 signaling pathways. Biochem Biophys Res Commun.
286:923–928. 2001.
|
|
24
|
Liu XH, Kirschenbaum A, Yao S, et al:
Upregulation of vascu larendothelial growth factor by cobalt
chloride-simulated hypoxia is mediated by persistent induction of
cyclooxygenase-2 in a metastatic human prostate cancer cell line.
Clin Exp Metastasis. 17:687–694. 1999.
|
|
25
|
Uefuji K, Ichikura T and Mochizuki H:
Cyclooxygenase-2 expression is related to prostaglandin
biosynthesis and angiogenes is in gastric cancer. Clin Cancer Res.
6:135–138. 2000.
|
|
26
|
Bamba H, Ota S, Kato A, et al:
Prostaglandins up-regulate vascu larendothelial growth factor
production distict pathways in differentiated U937 cells. Biochem
Biophys Res Commun. 273:487–491. 2000.
|
|
27
|
Tsurusaki T, Kanda S, Sakai H, et al:
Vascular endothelial growth factor-C expression in human prostatic
carcinoma and its relationship to lymph node metastasis. Br J
Cancer. 80:309–313. 1999.
|
|
28
|
Su JL, Shih JY, Yen ML, et al:
Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular
endothelial growth factor-C up-regulation: a novel mechanism of
lymph angiogenesis in lung adenocarcinoma. Cancer Res. 64:554–564.
2004.
|
|
29
|
Kubowicz P, Żelaszczyk D and Pękala E:
RNAi in clinical studies. Curr Med Chem. 20:1801–1816. 2013.
|
|
30
|
Ghafouri-Fard S and Ghafouri-Fard S: siRNA
and cancer immunotherapy. Immunotherapy. 4:907–917. 2012.
|
|
31
|
McCormick DL, Madigan MJ and Moon RC:
Modulation of rat mammary carcinogenesis by indomethacin. Cancer
Res. 45:1803–1808. 1985.
|
|
32
|
Harris RE, Alshafie GA, Abou-Issa H and
Seibert K: Chemoprevention of breast cancer in rats by celecoxib, a
cyclooxygenase 2 inhibitor. Cancer Res. 60:2101–2103. 2000.
|
|
33
|
Nakatsugi S, Ohta T, Kawamori T, et al:
Chemoprevention by nimesulide, a selective cyclooxygenase-2
inhibitor, of
2-amino-1-methyl-6-phenylimidazol[4,5-b]pyridine(PhIP)-induced
mammary gland carcinogenesis in rats. Jpn J Cancer Res. 91:886–892.
2000.
|
|
34
|
Harris RE, Chlebowski RT, Jackson RD, et
al: Women’s Health Initiative: Breast cancer and nonsteroidal
anti-imflammatory drugs: prospective results from the Women’s
Health Initiative. Cancer Res. 63:6096–6101. 2003.
|
|
35
|
Khuder SA and Mutgi AB: Breast cancer and
NSAIDs use: a meta-analysis. Br J Cancer. 84:1188–1192. 2001.
|
|
36
|
Jana D, Sarkar DK, Maji A, et al: Can
cyclo-oxygenase-2 be a useful prognostic and risk stratification
marker in breast cancer? J Indian Med Assoc. 110:429–433. 2012.
|