Introduction
Squamous cell carcinoma is the most common type of
malignant neoplasm of the oral mucosa and accounts for >90% of
all intraoral malignant tumors, in Korea (1). In Korea, ~20% head and neck cancer is
estimated to occur in the oral region (1). Despite gradual improvements in
surgery, radiotherapy and chemotherapy treatments, the prognosis of
patients presenting with locally advanced oral squamous cell cancer
(OSCC) remains poor, with only a 50% survival rate over five years
(2). Due to these poor survival
rates and the severe functional impairment caused by surgery and
radiation, the development of novel therapeutic strategies in the
management of patients with advanced OSCC is urgently required.
Head and neck SCC cell lines in cultures are widely
used to understand therapeutic development. However, the
establishment of SCC cell lines is considered difficult and low
success rates have been reported. Cell lines have previously been
derived from patients who had received radiotherapy, with or
without chemotherapy (3); however,
radiotherapy and chemotherapy eliminate sensitive cell populations
and may alter the in vivo progression and cell heterogeneity
of the tumor (4). Genetic
abnormalities in human cancer are markedly geographically
dependent, and the cultural and environmental background of the
patient are closely associated with the carcinogenic process. For
example, oral cancer has been clearly associated with the presence
of human papillomavirus HPV16 in Western countries, but not in
Korea (5). In the current study, YD
cell lines, which are newly established oral cancer cell lines
originating from untreated oral tumors in Korean patients, were
used (5). The YD cell lines were
derived from untreated primary tumors of the tongue (YD-8), buccal
mucosa (YD-9) and lower gingiva (YD-38), and the cell lines
exhibited genetically different p53 statuses. The YD-8 cell line
had a point mutation at codon 273 of exon 8, which is involved in
the DNA-binding site, revealing its significance in p53
transcriptional activation; the GGT (arginine) sequence was
replaced with CAT (histidine). This R273H mutation accounts for
~20% p53 missense mutations (6).
The YD-9 and YD-38 cells did not have the p53 mutation; however,
the p53 protein was positively expressed in the YD-9 cells but not
in the YD-38 cells. As over half of all human cancers lose p53
function through mutation (7),
investigation of the potential impact of p53 mutations on disease
pathology and therapeutic response is important. Tumors with an
inactive mutant p53 are aggressive and are commonly resistant to
ionizing radiation and chemotherapy (8).
DNA topoisomerase I (Top1), an essential nuclear
enzyme that controls and modifies the topological state of DNA in
numerous cellular metabolic processes (9,10),
serves as a target for screening anticancer agents (10–12).
CKD-602 (7-[2-(N-isopropylamino) ethyl]-(20S)-camptothecin;
belotecan), a Top1 inhibitor, is a novel, synthetic, water-soluble
camptothecin derivative (13).
Preclinical trials of CKD-602 have demonstrated that CKD-602 exerts
antitumor activity against various human tumor cell lines, and that
the results are equal or superior to those of camptothecin
(13). In a previous study, CKD-602
was observed to exert an in vitro anticancer effect on three
OSCC cell lines, A253 (submandibular gland), HSC-3 (tongue) and KB
(oral mucosa) (14). In the present
study, the potential effects of CKD-602 on cell viability in OSCC
cell lines originating from oral cancer in Korean patients with
genetically different p53 statuses was evaluated, as well as the
mechanisms underlying the induction of cell cycle arrest and
apoptosis.
Materials and methods
Reagents
CKD-602 (Chong Kun Dang Pharmaceutical Corp., Seoul,
Korea) was dissolved in distilled water at 1 μg/ml, and stored as a
stock solution in aliquots at −20°C until use. Final concentrations
between 0.01 and 10 μg/ml CKD-602 were obtained by appropriate
dilutions of the stock solution with RPMI 1640 medium (Gibco-BRL,
Grand Island, NY, USA).
Cell lines and cell culture
Three OSCC cell lines, YD-8 (60501; tongue), YD-9
(60502; buccal mucosa) and YD-38 (60508; lower gingiva) were used
(4). All cell lines were obtained
from the Korean Cell Line Bank (Seoul, Korea).
Each cell line was maintained in RPMI-1640 medium
(Gibco-BRL), supplemented with 10% heat-inactivated fetal bovine
serum (FBS, Gibco-BRL), 100 μg/ml streptomycin (Gibco-BRL) and 100
IU/ml penicillin (Gibco-BRL), as a monolayer under standard
conditions (37°C, and in a humidified atmosphere of 5%
CO2). To transfer or passage the cell lines, each
confluent monolayer was washed with phosphate-buffered saline (PBS;
Welgene, Daegu, Korea) and detached with a 0.05% trypsin/0.02% EDTA
solution (Gibco-BRL).
MTS viability assay
Cells at a density of 2×104 cells/well in
100 μl RPMI with 10% FBS were added to the wells of a 96-well
plate. The cells were treated with different concentrations (0.01,
0.1, 0.5, 1, 5 and 10 μg/ml) of CKD-602 for 24, 48 and 72 h.
Control samples of each cell line were treated with medium only.
For the viability assay, 20 μl/well CellTiter 96®
AQueous One Solution Reagent (MTS; Promega Corporation, Madison,
WI, USA) was added. After 1 h incubation at 37°C in a humidified
atmosphere of 5% CO2, the absorbance at 490 nm was
recorded using an ELISA plate reader (Bio-Tek Instruments, Inc.,
Winooski, VT, USA) The assay was performed in triplicate with three
independent experiments for each condition. The data from the
treatment groups were normalized to those of the control samples
and are presented as the mean ±standard error of the mean. The half
maximal (50%) inhibitory concentration (IC50) values
were calculated from the dose-response curve.
Annexin assay
Apoptosis was quantified using fluorescein
isothiocyanate (FITC)-Annexin V Apoptosis Detection kit I (BD
Biosciences, San Jose, CA, USA) according to the manufacturer’s
instructions. Briefly, the cells were plated at a density of
1×106 cells/well in a 100 mm culture dish, treated with
0.1 and 0.5 μg/ml CKD-602 for 48 h, and harvested by centrifugation
at 500 × g for 3 min. The cell pellets were resuspended in Annexin
V binding buffer containing 140 mM CaCl2, 10 mM
HEPES/NaOH and 2.5 mM MgCl2. FITC-conjugated Annexin V
(5 μl) and propidium iodide (PI; 5 μl) were added to the cells, and
the mixtures were incubated for 15 min at room temperature in the
dark. The analyses were performed using a Fluorescence-Activated
Cell Sorting (FACScan) instrument (Becton-Dickinson, Franklin
Lakes, CA, USA). Annexin V- and PI-positive cells were considered
to be apoptotic.
Cell cycle analysis
The cells were plated at a density of
1×106 cells/well in 100 mm culture dishes. The cells
were treated with 0.02 μg/ml CKD-602 for 48 h, harvested by
centrifugation at 500 × g for 3 min, washed twice in ice-cold PBS,
fixed in 70% ethanol and stored at −20°C for a minimum of 1 h;
subsequently, cells were washed with ice-cold PBS and resuspended
in 500 μl PI/RNase Staining Buffer (BD Biosciences). The cell cycle
position was evaluated by FACScan using an excitation laser set at
480 nm and a detection wavelength of 575 nm. A minimum of 10,000
events/sample was analyzed.
Western blot analysis
CKD-602-treated and non-treated cells were suspended
in RIPA buffer (Rockland, Gilbertsville, PA, USA) containing 5 μM
AEBSF, 1.5 μM aprotinin, 10 μM E-64, 0.01 μM leupeptin and
phosphatase inhibitors [1 mM sodium orthovanadate
(Na2VO4; S6508; 1 mM sodium molybdate
(Na2MoO4; M1003) 4 mM sodium tartrate
dihydrate (S4797); 2 mM imidazole (I0125) all purchased from
Sigma-Aldrich, (St. Louis, MO, USA)], and were placed on ice for 20
min. Following centrifugation at 4°C at 10,000 × g for 20 min, the
cell supernatant was collected. The protein concentration was
determined using a bicinchoninic protein assay kit (Pierce,
Rockford, IL, USA). The whole lysate (20 μg) was resolved on a 10%
or 12.5% SDS-PAGE gel, transferred to a polyvinylidene difluoride
membrane (Bio-Rad, Hercules, CA, USA) by electroblotting, and
probed with polyclonal rabbit anti-human β-actin (4967), monoclonal
mouse anti-human p53 (9826), monoclonal mouse anti-human phospho-H3
(Ser 10; 3377), polyclonal rabbit anti-human cyclin B1 (4138),
monoclonal rabbit anti-human cyclin A2 (4656), polyclonal rabbit
anti-human cdc2 (9112), polyclonal rabbit anti-human phopho-cdc2
(Tyr 15; 9111), polyclonal rabbit anti-human Myt1 (4282) or
polyclonal rabbit anti-human phospho H2AX (Ser 130; 9718)
antibodies (1:1,000; Cell Signaling Technology, Inc. (Danvers, MA,
USA) overnight at 4°C. The membrane was washed three times with 1×
PBS and 0.05% Tween-20 for 15 min, and incubated for 1 h with a
horseradish peroxidase-conjugated polyclonal horse anti-rabbit
(7074) or polyclonal goat anti-mouse (7076) antibody. (1:2,000;
Cell Signaling Technology, Inc.). The blot was developed using an
enhanced chemiluminescence kit (Intron Biotechnology Inc.,
Seongnam, Korea).
Results
Effects of CKD-602 on cell viability of
OSCC cell lines
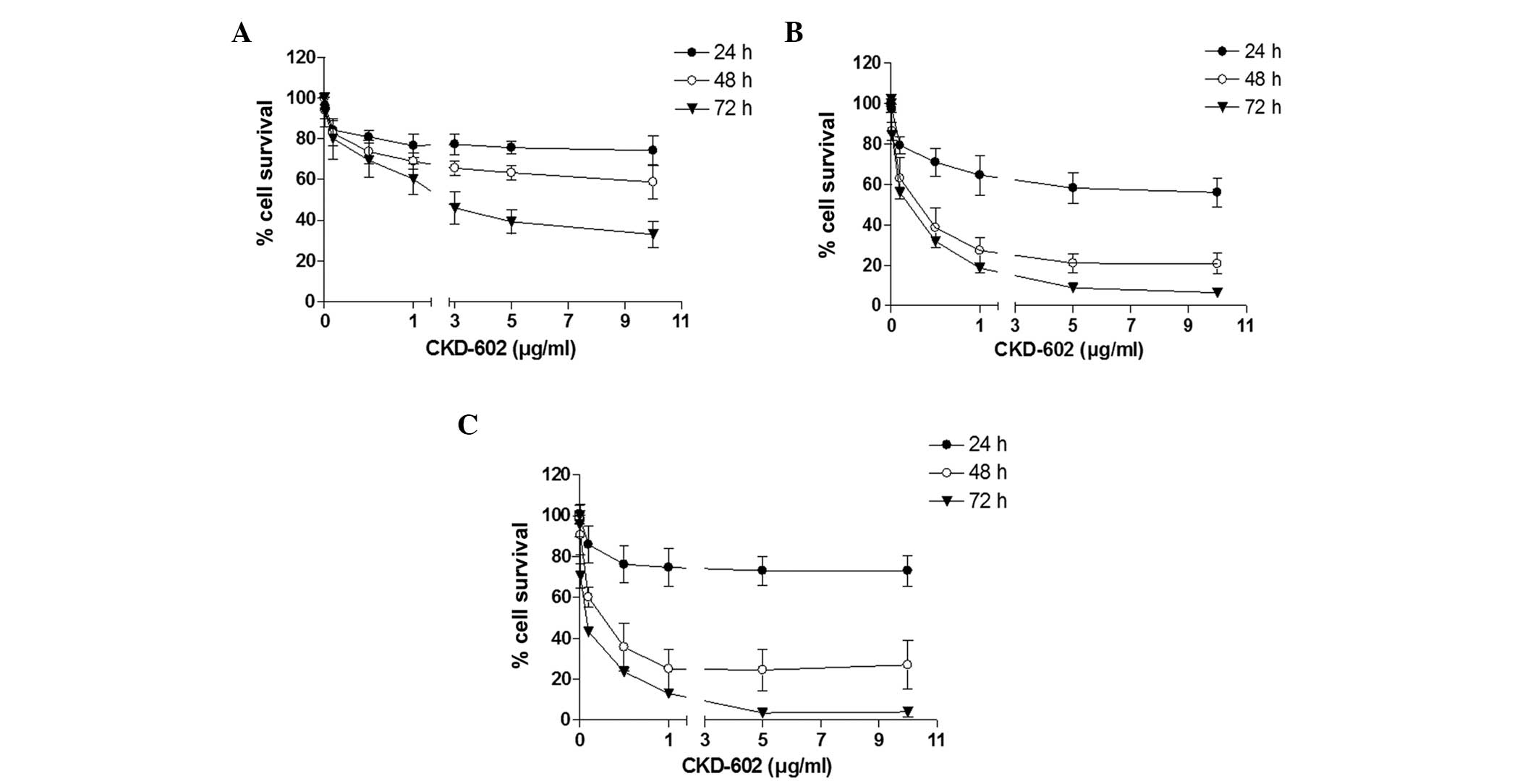
Using the cell viability assay, CKD-602 was revealed
to exert a significant cytotoxic effect on all cell lines in a
time- and dose-dependent manner (Fig.
1). The cell viability IC50 values were 2.4 μg/ml
for YD-8, 0.18 μg/ml for YD-9 and 0.05 μg/ml for YD-38 cells at 72
h following treatment (Table
I).
 | Table IGrowth inhibition (IC50,
μg/ml) of oral squamous cell cancer cell lines by CKD-602. |
Table I
Growth inhibition (IC50,
μg/ml) of oral squamous cell cancer cell lines by CKD-602.
| IC50,
μg/ml |
|---|
|
|
|---|
| Treatment
duration | YD-8 | YD-9 | YD-38 |
|---|
| 48 h | - | ≥0.3 | ≥0.24 |
| 72 h | ≥2.4 | ≥0.18 | ≥0.05 |
Effects of CKD-602 on the OSCC cell
cycle
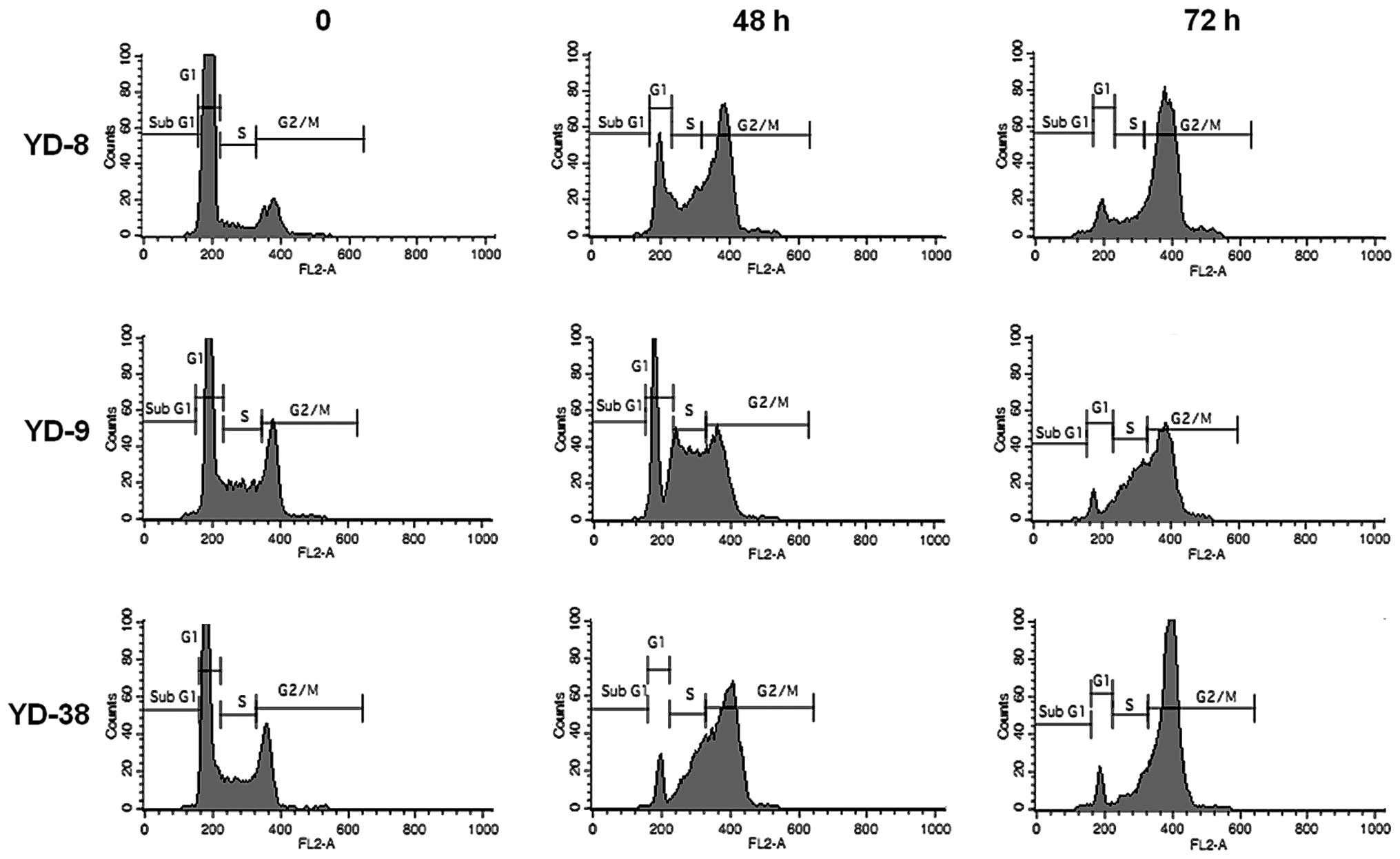
Cell cycle analysis was performed following
treatment with 0.02 μg/ml CKD-602 for 48 h and 72 h in each cell
line. CKD-602 induced G2/M phase cell accumulation in all cell
lines (Fig. 2). The proportion of
the cell population in G2/M phase at 72 h increased from 11.9±3.8
to 77.6±0.3% for YD-8 cells, from 25.2±3.6 to 54.0±5.4% for YD-9
cells and from 19.6±3.5 to 78.3±2.6% for YD-38 cells. The
percentage of the cell population in the G1 phase at 72 h was
reduced from 81.7±6.48 to 10.9±2.0% for YD-8 cells, from 49.4±4.5
to 7.3±1.56% for YD-9 cells and from 58.8±7.0 to 9.0±2.9% for YD-38
cells.
Effects of CKD-602 on cell cycle
regulatory protein expression levels
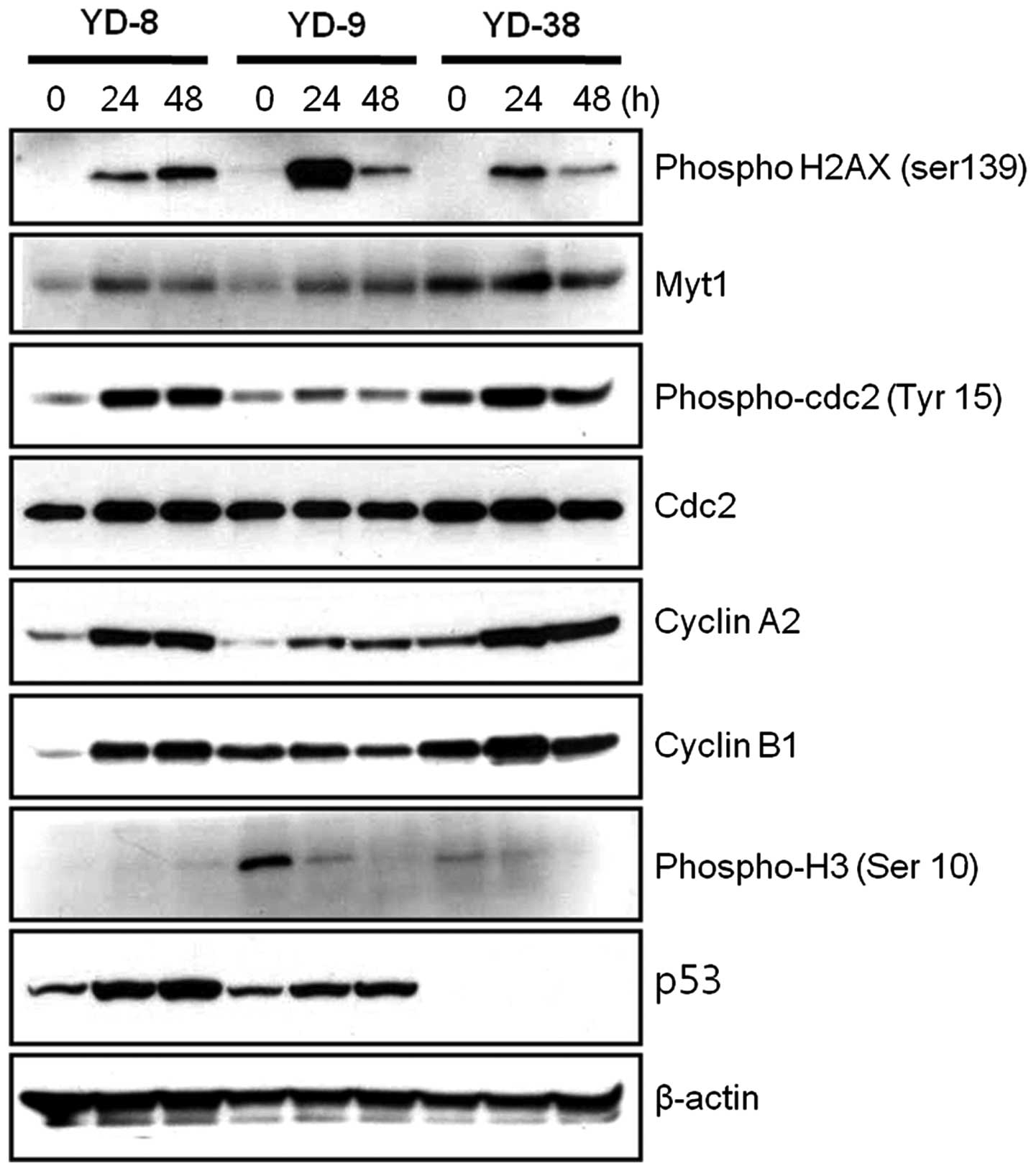
As CKD-602 induced G2/M phase arrest in the OSCC
cell lines, whether alterations in the cell cycle regulatory
proteins occurred following treatment with a non-cytotoxic dose of
CKD-602 (0.02 μg/ml) was subsequently investigated. The
phosphorylation of histone H2AX (γH2AX) Ser 139 is one of the early
events initiated by double-stranded DNA breaks. Immunoblotting
analysis revealed that the phosphorylation of histone H2AX was
increased in the CKD-602 treatment cells. In the cell cycle,
distinct cyclin/cyclin-dependent kinase complexes are activated to
regulate cell cycle progression. Cyclin A- and cyclin B-associated
cdc2 regulates the G2/M phases. During G2 phase, the cdc2/cyclin B
complex is maintained as inactive by the phosphorylation of cdc2
Tyr 15 and Thr 14 by the Myt1 kinases (15–17).
As shown in Fig. 3, the Myt1
protein expression levels in the three YD cell lines were observed
to be increased at 24 h and reduced at 48 h CKD-602 treatment. The
expression of cdc2, which is involved in cell cycle arrest in the
G2 phase, was not significantly affected by CKD-602 as compared
with the control treatment, however, the phosphorylated form,
phospho-cdc2 (Tyr 15), was significantly increased at 24 h in all
three YD cell lines, and reduced at 72 h in the YD-9 and YD-38
cells (P<0.05). Cyclin A2 protein expression levels were
increased at 24 h and 48 h in the three YD cell lines subsequent to
CKD-602 treatment as compared with the controls. Cyclin B1 protein
expression levels were increased at 24 h following CKD-602
incubation in YD-8 and YD-38 cells, however, no change at 24 h was
observed in the YD-9 cell line. These increases in cyclin B1
expression levels were reduced at 48 h following CKD-602 treatment
in the YD-9 and YD-38 cell lines. p53 protein expression was
detected by immunoblotting in the YD-8 and YD-9 cells, but not in
the YD-38 cells (Fig. 3).
As the phosphorylation of histone H3 is a molecular
checkpoint for entering mitosis (18), the phosphorylation of histone H3 Ser
10 following CKD-602 treatment was examined. As shown in Fig. 3, the phosphorylation of histone H3
Ser 10 was significantly reduced in a time-defendant manner in the
YD-9 and YD-38 cell lines following CKD-602 treatment (P<0.05),
however, this was not detected in the YD-8 cell line.
Effects of CKD-602 on apoptosis in OSCC
cell lines
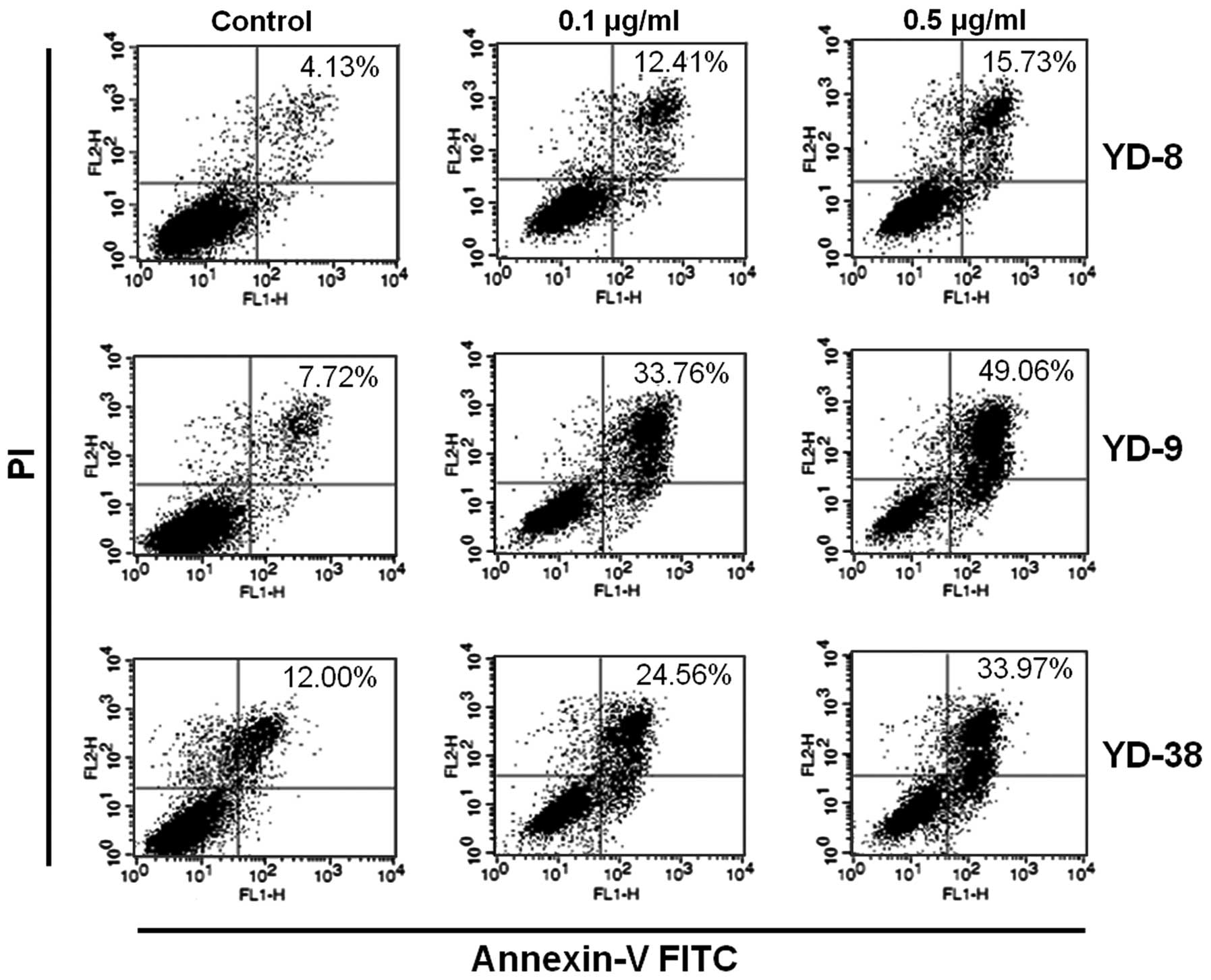
Annexin V/FITC staining, along with flow cytometry,
enables the quantitative assessment of living (Annexin
V-FITC-negative/PI-negative), early apoptotic (Annexin
V-FITC-positive/PI-negative), late apoptotic/necrotic (Annexin
V-FITC-positive/PI-positive) and dead (Annexin
V-FITC-negative/PI-positive) cells. The effects of 24 h CKD-602
treatment on YD cell apoptosis are shown in Fig. 4 and Table II. The cells in the lower right
quadrant indicate early apoptosis and the cells in the upper right
quadrant signify late apoptosis. The proportion of
Annexin-V/FITC-staining cells was markedly increased in a
dose-dependent manner following treatment with CKD-602. However,
the apoptotic proportion was lower in the YD-8 cells than in the
YD-9 or YD-38 cell lines.
| Table IIResults of FACS analysis evaluating
apoptotic effects following treatment with CKD-602 for 48 h. |
Table II
Results of FACS analysis evaluating
apoptotic effects following treatment with CKD-602 for 48 h.
| Fraction of cells,
% |
|---|
|
|
|---|
| YD-8 | YD-9 | YD-38 |
|---|
|
|
|
|
|---|
| CKD-602 | Live | Apoptotic | Live | Apoptotic | Live | Apoptotic |
|---|
| Control | 94.06±0.13 | 4.78±0.91 | 92.14±1.91 | 6.57±1.63 | 88.03±5.04 | 7.15±3.62 |
| 0.1 μg/ml | 87.97±3.80 | 10.24±3.07 | 67.58±4.12 | 30.91±4.04 | 82.56±6.87 | 14.56±3.96 |
| 0.5 μg/ml | 83.73±4.19 | 13.77±2.77 | 60.16±5.46 | 38.74±14.6 | 70.85±3.51 | 26.31±1.20 |
Discussion
Topoisomerase inhibitors are a class of agents that
target topoisomerase specifically by intercalating inside the
topoisomeares cleavage complexes and mediate the changes in DNA
structure during the normal cell cycle. Recently, topoisomerases
have become widely investigated targets for cancer chemotherapy
treatment. Topoisomerase inhibitors are hypothesized to suppress
the regulatory step, which normally reseals the parent strand of
DNA following passage of the daughter strand (19). The collision of the replication fork
with the cleaved strand of DNA causes an irreversible
double-stranded DNA break, which arrests the process of cell
division and results in cell death (19).
CKD-602 is a potent Top1 inhibitor that successfully
overcomes the poor water solubility and toxicity of the
corresponding parent drug, camptothecin. Clinical trials for the
treatment of various types of cancer with CKD-602 are ongoing and
have shown promising results (13,14,20,21).
CKD-602 may have potential in the treatment of patients with oral
squamous cell carcinoma, as determined by the potent anticancer
effects of the drug (14).
In the present study, CKD-602 induced cytotoxicity
in OSCC cell lines, causing apoptosis in a dose- and time-dependent
manner. The cytotoxic effect of CKD-602 was more prominent in the
YD-9 and YD-38 cell lines, which did not possess p53 mutations,
than in the YD-8 cell line, which did have a p53 mutation (Fig. 1 and Table I). Furthermore, the YD-9 and YD-38
cell lines exhibited more prominent apoptosis than the YD-8 cell
line following CKD-602 treatment (Fig.
4 and Table II). Conflicting
data have been reported concerning the influence of the p53 gene on
the efficacy of topoisomerase inhibitors. Wang et al
(22) demonstrated that a p53
disruption sensitizes glioblastoma cells to Top1 inhibitor-mediated
apoptosis and wild-type p53 promotes a senescence-like phenotype,
subsequent to SN-38 treatment. In another study, the cytotoxic
effect of CKD-602 was more prominent in mutant p53 cell lines than
in wild-type p53 cell lines (20).
The involvement of p53 in the cytotoxic effects of anticancer
agents remains under debate. Conflicting results have been
generated using genetic models where p53 function has been modified
by E6 protein expression (22,23) or
homologous recombination (24), as
cells with non-functional p53 may develop greater sensitivity,
greater resistance or retain the same sensitivity, depending on the
drug administered and the cellular context. Recent studies have
refuted the role of p53 in determining differential
susceptibilities to Top1 inhibitor (25,26).
As determined by this evidence, the association between the p53
status and the effect of Top1 inhibitors, including CKD-602,
requires further evaluation.
As over half of human tumors exhibit a p53 mutation
or deficiency, the investigation of cell cycle checkpoints in tumor
cells with various p53 statuses provides a potential basis for
developing novel tumor therapeutics. In the present study, three
OSCC cell lines were used with genetically different p53 statuses.
The YD-8 cell line had a point mutation at codon 273 of exon 8 and
the GGT sequence was altered to CAT, which resulted in a change
from arginine to histidine. The levels of p53 protein were detected
in YD-8 and YD-9 by immunoblotting (Fig. 3). As the half-life of wild type p53
protein is several minutes, p53 protein levels in normal cells are
relatively low and are generally undetectable by immunoblotting. An
abnormal p53 protein may be easily detected by immunoblotting due
to the prolongation of half-life. However, the YD-9 cell line,
which did not exhibit the p53 mutation, positively expressed the
p53 protein (Fig. 3). Several
possible underlying mechanisms, other than a point mutation, may
result in the overexpression of the p53 protein (27,28).
For example, genetic alternations in another region of the exon,
such as the promoter or the intron of the p53 gene, may result in
higher expression levels of the wild-type p53. The YD-38 cell line
did not possess a p53 gene mutation and the p53 protein was not
detected by immunoblotting (Fig.
3).
Treatment of the cells with CKD-602 for 48 h and 72
h resulted in cell cycle arrest at the G2/M phase (Fig. 2). This effect was associated with
alterations in the expression of cyclins, including cyclin A and
cyclin B1, molecules which provide molecular determinants for the
cell cycle. This finding coincides with those from other studies
with regard to the effect of CKD-602 on glioma cell lines. Kim
et al (20) reported a
reduction in the percentage of cells in the G1 phase and an
increase in the percentage of cells in the G2/M phases in the U87
MG, U343 MG, U251 MG and LN229 human glioma cell lines following
treatment with CKD-602.
In conclusion, in the present study, CKD-602 was
demonstrated to exert an in vitro anticancer effect against
OSCC cell lines by promoting cell cycle arrest in the G2/M phase
and by inducing apoptosis. These findings suggest that CKD-602 is a
promising candidate for use in oral cancer therapy and provides a
rationale for the further evaluation of CKD-602 treatment in oral
cancer by in vivo and clinical studies.
Acknowledgements
This study was supported by the SNUBH Research Fund
(grant no. 02-2011-001).
References
|
1
|
Kim KM, Kim YM, Shim YS, et al: Study
Group of Korean Society of Head and Neck Surgerons: Epidemiologic
survey of head and neck cancers in Korea. J Korean Med Sci.
18:80–87. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM: Global cancer statistics in the
year 2000. Lancet Oncol. 2:533–543. 2001. View Article : Google Scholar
|
|
3
|
Easty DM, Easty GC, Carter RL, Monaghan P
and Butler LJ: Ten human carcinoma cell lines derived from squamous
carcinomas of the head and neck. Br J Cancer. 43:772–785. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nicolson GL: Tumor progression, oncogenes
and the evolution of metastatic phenotypic diversity. Clin Exp
Metastasis. 2:85–105. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee EJ, Kim J, Lee SA, et al:
Characterization of newly established oral cancer cell lines
derived from six squamous cell carcinoma and two mucoepidermoid
carcinoma cells. Exp Mol Med. 37:379–390. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scheffner M, Werness BA, Huibregtse JM, et
al: The E6 oncoprotein encoded by human papillomavirus types 16 and
18 promotes the degradation of p53. Cell. 63:1129–1136. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hainaut P, Hernandez T, Robinson A, et al:
IARC Database of p53 gene mutations in human tumors and cell lines:
updated compilation, revised formats and new visualisation tools.
Nucleic Acids Res. 26:205–213. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tagscherer KE, Fassl A, Sinkovic T, Combs
SE and Roth W: p53-dependent regulation of Mcl-1 contributes to
synergistic cell death by ionizing radiation and the Bcl-2/Bcl-XL
inhibitor ABT-737. Apoptosis. 17:187–199. 2012. View Article : Google Scholar
|
|
9
|
Champoux JJ: DNA topoisomerases:
structure, function, and mechanism. Annu Rev Biochem. 70:369–413.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pommier Y, Leo E, Zhang H and Marchand C:
DNA topoisomerases and their poisoning by anticancer and
antibacterial drugs. Chem Biol. 17:421–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gupta M, Fujimori A and Pommier Y:
Eukaryotic DNA topoisomerases I. Biochim Biophys Acta. 1262:1–14.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang JC: Cellular roles of DNA
topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol.
3:430–440. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee JH, Lee JM, Kim JK, et al: Antitumor
activity of 7-[2-(N-isopropylamino)ethyl]-(20S)-camptothecin,
CKD602, as a potent DNA topoisomerase I inhibitor. Arch Pharm Res.
21:581–590. 1998. View Article : Google Scholar
|
|
14
|
Ok YJ, Myoung H, Kim YK, et al: Apoptotic
effect of CKD-602 (Camtobell) on oral squamous cell carcinoma cell
lines. Oral Oncol. 45:266–272. 2009. View Article : Google Scholar
|
|
15
|
Booher RN, Holman PS and Fattaey A: Human
Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not
Cdk2 activity. J Biol Chem. 272:22300–22306. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu F, Stanton JJ, Wu Z and Piwnica-Worms
H: The human Myt1 kinase preferentially phosphorylates Cdc2 on
threonine 14 and localizes to the endoplasmic reticulum and Golgi
complex. Mol Cell Biol. 17:571–583. 1997.PubMed/NCBI
|
|
17
|
Parker LL and Piwnica-Worms H:
Inactivation of the p34cdc2-cyclin B complex by the human WEE1
tyrosine kinase. Science. 257:1955–1957. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Van Hooser A, Goodrich DW, Allis CD,
Brinkley BR and Mancini MA: Histone H3 phosphorylation is required
for the initiation, but not maintenance, of mammalian chromosome
condensation. J Cell Sci. 111:3497–3506. 1998.PubMed/NCBI
|
|
19
|
Holden JA: DNA topoisomerases as
anticancer drug targets: from the laboratory to the clinic. Curr
Med Chem Anticancer Agents. 1:1–25. 2001. View Article : Google Scholar
|
|
20
|
Kim YY, Park CK, Kim SK, et al: CKD-602, a
camptothecin derivative, inhibits proliferation and induces
apoptosis in glioma cell lines. Oncol Rep. 21:1413–1419.
2009.PubMed/NCBI
|
|
21
|
Lee JH, Lee JM, Lim KH, et al: Preclinical
and phase I clinical studies with Ckd-602, a novel camptothecin
derivative. Ann N Y Acad Sci. 922:324–325. 2000. View Article : Google Scholar
|
|
22
|
Wang Y, Zhu S, Cloughesy TF, Liau LM and
Mischel PS: p53 disruption profoundly alters the response of human
glioblastoma cells to DNA topoisomerase I inhibition. Oncogene.
23:1283–1290. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gupta M, Fan S, Zhan Q, et al:
Inactivation of p53 increases the cytotoxicity of camptothecin in
human colon HCT116 and breast MCF-7 cancer cells. Clin Cancer Res.
3:1653–1660. 1997.
|
|
24
|
Bunz F, Dutriaux A, Lengauer C, et al:
Requirement for p53 and p21 to sustain G2 arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Djuzenova CS, Güttler T, Berger S, Katzer
A and Flentje M: Differential response of human glioblastoma cell
lines to combined camptothecin and ionizing radiation treatment.
Cancer Biol Ther. 7:364–373. 2008. View Article : Google Scholar
|
|
26
|
Morandi E, Severini C, Quercioli D, et al:
Gene expression time-series analysis of camptothecin effects in
U87-MG and DBTRG-05 glioblastoma cell lines. Mol Cancer. 7:662008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wong RH, Du CL, Wang JD, et al: XRCC1 and
CYP2E1 polymorphisms as susceptibility factors of plasma mutant p53
protein and anti-p53 antibody expression in vinyl chloride
monomer-exposed polyvinyl chloride workers. Cancer Epidemiol
Biomarkers Prev. 11:475–482. 2002.PubMed/NCBI
|
|
28
|
Yook JI and Kim J: Expression of
p21WAF1/CIP1 is unrelated to p53 tumour suppressor gene status in
oral squamous cell carcinomas. Oral Oncol. 34:198–203. 1998.
View Article : Google Scholar : PubMed/NCBI
|