Introduction
Histone acetylation is associated with the genesis
and development of certain tumors and is regulated by histone
acetyltransferase (HAT) and histone deacetylase (HDAC) (1,2). Thus,
suppressing HDAC can be used as a novel antitumor therapy (3,4). HDAC
inhibitors (HDACIs) are notable due to their antitumor function
(5,6). However, numerous HDACIs that are
currently used in the clinic, including trichostatin A (TSA),
apicidin and suberoylanilide hydroxamic acid (SAHA), have been
restricted due to toxicity and a short half-life (7). Valproate acid sodium (VPA), a
short-chain fatty acid with the chemical name 2-sodium valproate,
was demonstrated to be a specific HDAC inhibitor and has been used
widely as an anticonvulsant drug with low toxicity and a long
half-life (8).
Classical therapy for hepatocellular carcinoma, a
malignant tumor that exhibits a quick progression, poor prognosis
and high mortality rate, is unsatisfactory and novel treatment
methods are required (9). In the
present study, VPA was used to reverse the malignant phenotypes of
hepatocellular carcinoma through regulating the level of histone
acetylation, and the HDACI mechanism of VPA was determined. The
apoptosis pathway of hepatocellular carcinoma HepG2 cells was also
identified and, finally, the anticarcinoma effects of VPA on a
hepatocellular carcinoma mouse model were estimated in
vivo.
Materials and methods
Cell culture and induction
HepG2, BEL-7402 and SMMC-7721 cells (Cell Bank of
Type Culture Collection of Chinese Academy of Sciences, Shanghai,
China) were cultured in RPMI-1640 standard medium (Gibco Life
Technologies) supplemented with 10% fetal bovine serum (Tianhang,
Zhejiang, China), glutamine (Tianhang) and antibiotics (50 IU
penicillin and 50 μg/ml streptomycin; Sigma-Aldrich, St. Louis, MO,
USA) in a humidified 5% CO2 and air atmosphere at 37°C.
Exponentially growing HepG2 cells were incubated in six-well plates
at a concentration of 1×105/ml. Subsequent to culturing
at 37°C in 5% CO2 for 2 h, 3.0 mmol/l VPA
(Sigma-Aldrich) was added. After a 48-h induction, the cells were
harvested for the following experiments.
Effect of VPA on HDAC activity and gene
expression
HDAC activity
TheHepG2, BEL-7402 and SMMC-7721 cells
(5×104 /ml) were induced by 3.0 mmol/l VPA for 48 h. The
cells were collected and 100 μg nuclear extract was used to detect
the total HDAC activity using a colorimetric HDAC activity assay
kit (BioVision, Inc., Milpitas, CA, USA), according to the
manufacturer’s instructions.
mRNA expression of HDAC1
HDAC1 mRNA expression was detected by reverse
transcription-polymerase chain reaction (RT-PCR). Total RNA was
extracted from the cells using TRIzol reagent (Gibco Life
Technologies, Carlsbad, CA, USA) and RT-PCR was performed. The PCR
products were assayed by 1% agarose gel electrophoresis, visualized
under a gel-image analysis system (Uvitec Ltd., Cambridge,
Cambridgeshire, UK) and then analyzed using the UVIband image
analyzer (Uvitec Ltd.). The relative intensity of objective HDAC1
mRNA was indicated by the ratio of the objective optical density
(OD) to the OD for β-actin. The control cells were treated with the
culture medium without VPA.
Cell culture and proliferation assay
Exponentially growing HepG2, BEL-7402 and SMMC-7721
cells (0.1 ml) were incubated in 96-well plates at a concentration
of 1×105 cells/ml. Subsequent to culturing at 37°C in 5%
CO2 for 2 h, 3.0 mmol/l VPA was added. After 48 h, the
cell proliferation was assessed by 10 μl MTT (5 mg/ml). The control
cells were cultured without VPA. The cell growth inhibition rate
(%) was calculated as follows: (Acontrol cell −
AVPA-treated cell) / Acontrol cell × 100,
where ‘A’ is the absorbance.
Cell cycle assay and expression of
associated gene and protein
Cell cycle assay
In accordance with the assay results, the HepG2 cell
line was used in the following detection. The HepG2 cells were
induced using the aforementioned method with 3.0 mmol/l VPA for 48
h. The cells were then collected and washed twice in
phosphate-buffered saline (PBS) and incubated overnight in cold 70%
ethanol at 4 °C. Subsequently, the cells were rinsed with PBS and
stained with propidium iodide (PI) working solution (0.2 mg/ml PI,
0.08 mg/ml ribonuclease A and 0.5 mg/ml trypsin inhibitor; Sigma)
for 30 min at room temperature and in the dark. The stained nuclei
were analyzed using flow cytometry (FCM; Beckman Coulter, Brea, CA,
USA) and the cell cycle was analyzed by the MacCycle software
(Beckman Coulter). The control cells were cultured without VPA.
mRNA expression of cyclins and
P21Waf/cip1
HepG2 cells were induced and RT-PCR was performed
using the aforementioned method to detect the expression of cyclin
A, D1 and E and P21Waf/cip1.
Protein expression of cyclins and
P21Waf/cip1
FCM was used to detect the protein expression of
cyclins A, D1 and E and P21Waf/cip1. The primary
antibodies used in this study were against cyclin A (mouse
monoclonal anti-cyclin A antibody, 6E6; cat. no. MS-1062-S0,-S1;
NeoMarkers, Fremont, CA, USA), cyclin D1 (mouse monoclonal
anti-cyclin D1 antibody, DCS-6; cat. no. MS-210-P0,-P1;
NeoMarkers), cyclin E (mouse monoclonal anti-cyclin E antibody,
HE12; cat. no. MS-870-P0,-P1; NeoMarkers) and
P21Waf/cif1 (mouse monoclonal anti-P21 antibody, F-5;
cat. no. SC-6246; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA). The monoclonal antibodies were diluted (1:40) using PBS
solution with 0.1% sodium azide. The secondary antibody was
FITC-rabbit polyclonal anti-mouse IgG (1:40; H+L; Signalway
Antibody, College Park, MD, USA). Briefly, 5×106 HepG2
cells were collected and washed following exposure to 3.0 mmol/l
VPA for 48 h. The cells were mixed with 1,000 μl permeabilization
buffer and incubated for 15 min at room temperature. The
supernatant liquor was replaced by 100 μl permeabilization buffer
following centrifugation at 2,000 × g for 10 min, and the cells
were suspended and mixed with 5 μl (1 μg) monoclonal antibodies for
cyclins A, D1 and E and P21Waf/cip1. After 30 min, the
cells were washed twice with PBS. The cells were then mixed with
100 μl (2.5 μg) secondary antibody at room temperature and in the
dark for 30 min. Subsequent to the cells being washed twice and
resuspended in PBS, protein expression was analyzed by FCM and the
mean fluorescence intensity exponent (MFI) was calculated.
Effect on HepG2 cell apoptosis and
caspases by VPA
Apoptosis assay
VPA (3.0 mmol/l) was used to induce HepG2 cell
apoptosis. Briefly, following 48 h of induction, the cells were
collected, washed once with PBS, stained with Annexin V/PI
according to the manufacturer’s instructions, and then analyzed
using FCM (Beckman Coulter) and MacCycle software. The control
cells were treated with the culture medium without VPA.
Caspase activity
To determine the pathway through which HepG2 cell
apoptosis is induced by VPA, the activity of caspases 3, 8 and 9
was measured using the caspase activity detection kit (Nanjing
KeyGen Biotech Co., Ltd., Nanjing, China). Briefly, HepG2 cells
were induced and collected, and were washed once using PBS. Protein
was extracted from the cells using cell lysates contained within
the kit, and the activity of caspases 3, 8 and 9 was measured
according to the manufacturer’s instructions. A405 was
used to denote caspase activity.
Caspase blocking assay
Caspase inhibitors were used to block caspase
expression. Briefly, HepG2 cells were induced by 3.0 mmol/l VPA
combined with 40 μmol/l caspase 3 inhibitor Z-DEVD-FMK, caspase 8
inhibitor Z-IETD-FMK and caspase 9 inhibitor Z-LEHD-FMK,
respectively (BioVision, Inc.). Apoptosis was detected using the
aforementioned method following 48 h. Cells induced by VPA alone
were used as the positive control, and normal control cells were
cultured with culture medium.
Protein expression of caspases
The protein expression of caspases 3, 8 and 9 was
detected using the aforementioned FCM technique.
Statistical analysis
Each experiment was performed at least in
triplicate. All the results were expressed as the mean ± standard
deviation. The data were analyzed by the Student’s unpaired t-test.
P<0.05 and P<0.01 were considered to indicate a statistically
significant difference.
Results
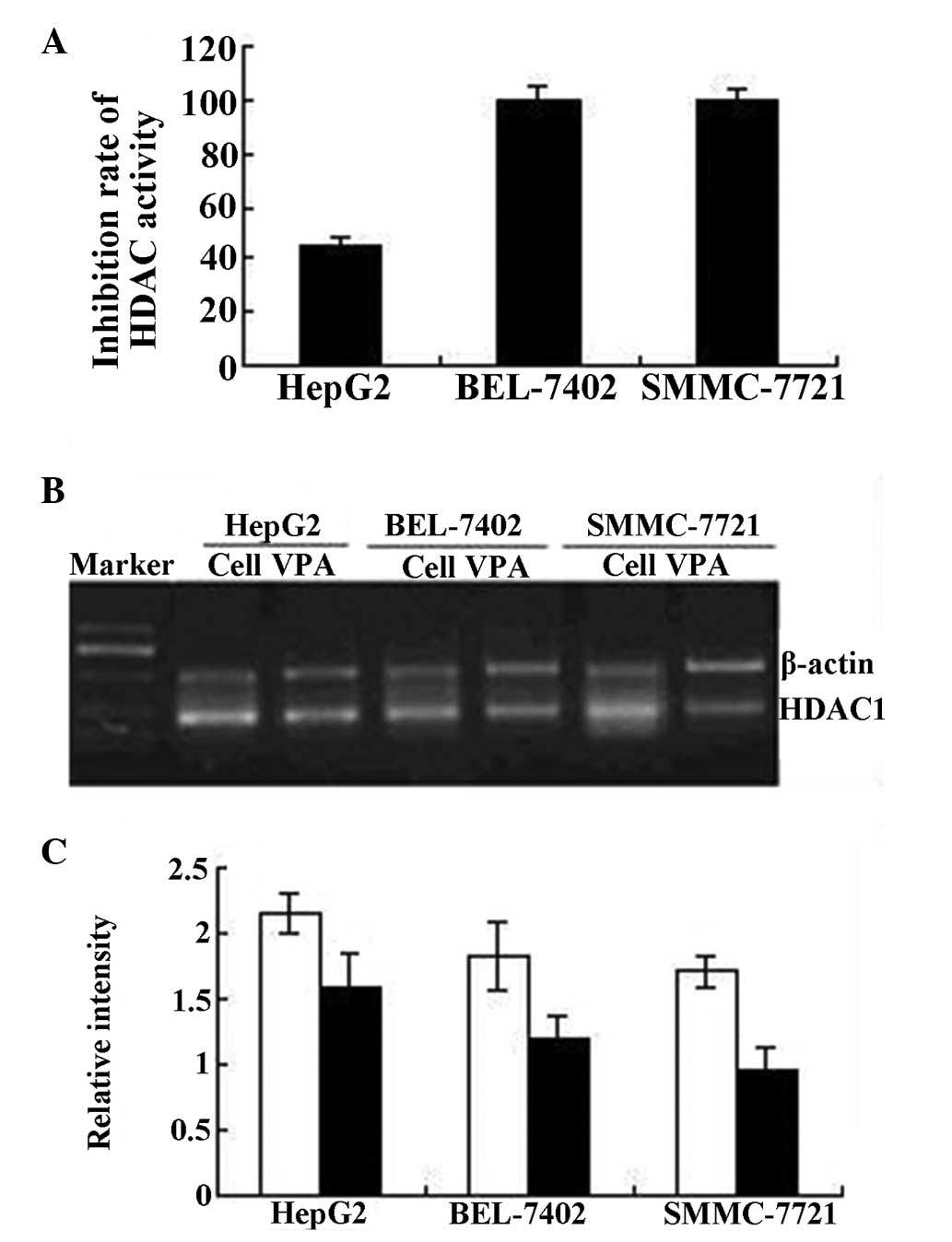
Effect of VPA on HDAC activity and gene
expression
The total HDAC activity of HepG2, BEL-7402 and
SMMC-7721 cells was markedly inhibited following 48 h of treatment
with VPA. HDAC activity in the BEL-7402 and SMMC-7721 cells was
completely inhibited (100 and 99.5%, respectively) by 3.0 mmol/l
VPA, while 45.1% of HDAC activity in HepG2 cells was inhibited. The
RT-PCR results revealed that the expression of HDAC1 mRNA in HepG2,
BEL-7402 and SMMC-7721 cells was also inhibited by VPA, as
described in Fig. 1.
Effect of VPA on the proliferation of
HepG2, BEL-7402 and SMMC-7721 cells
Compared with the control cells, the proliferation
of HepG2, BEL-7402 and SMMC-7721 cells was evidently inhibited by
3.0 mmol/l VPA, with an inhibition rate of 41.6, 62.6 and 69.8%,
respectively (Fig. 2).
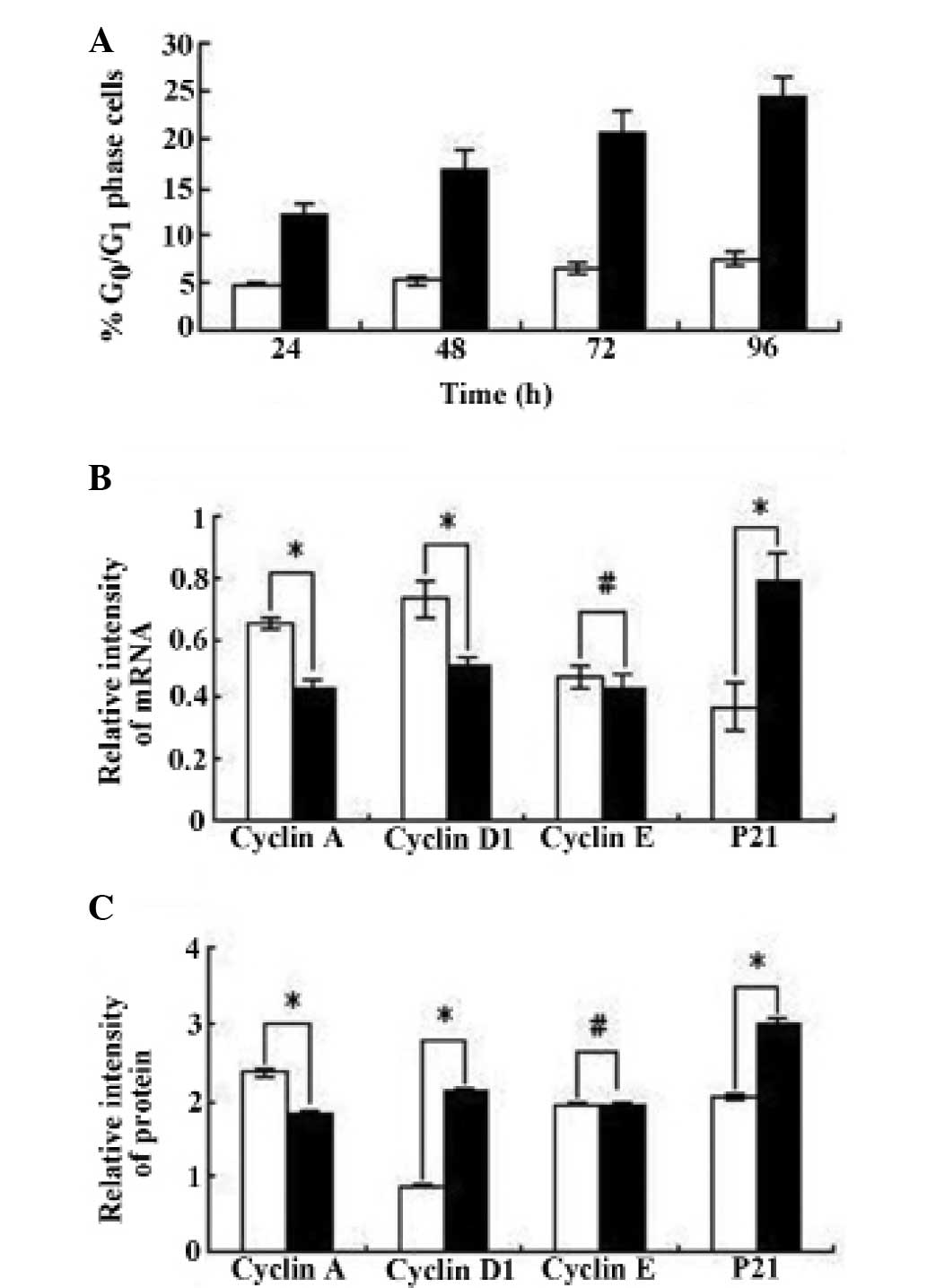
Effect of VPA on the cell cycle of HepG2
cells
Cell cycle profile
According to the inhibition of cell proliferation
and the cell cycle profile of HepG2 cells revealed by FCM assay,
there was no evident change in any phase of the cell cycle in the
control cells, but there was an increased proportion of VPA-treated
cells in the G0/G1 phase (P<0.01; Fig. 3A).
mRNA expression of cyclins A, D1 and
E, and P21Waf/cip1
mRNA expression was detected by RT-PCR assay.
Fig. 3B shows that compared with
the control cells, the mRNA expression of cyclins A and D1 was
downregulated and the expression of P21Waf/cip1 was
markedly upregulated in VPA-treated cells (P<0.01). However, the
expression of cyclin E was not significantly affected by VPA
(P>0.05).
Protein expression of cyclins A, D1
and E and P21Waf/cip1
Fig. 3C shows that
the protein expression detected by the FCM assay was in accordance
with the mRNA expression. The protein expression of cyclins A and
D1 was downregulated and P21Waf/cip1 expression was
markedly upregulated in cells incubated with VPA (P<0.01).
However, cyclin E expression demonstrated no significant difference
between the VPA-treated and control cells (P>0.05).
Effect on apoptosis of HepG2 cells by
VPA
HepG2 cell apoptosis induced by
VPA
Fig. 4A shows that,
as a HDAC inhibitor, the mechanism of proliferation inhibition
exerted by VPA on HepG2 cells was detected. Compared with the
control cells, the HepG2 cells treated with 3.0 mmol/l VPA for 48 h
demonstrated evident apoptosis and the apoptosis rate increased
from 6.25 to 29.5% (P<0.01).
 | Figure 4HepG2 apoptosis assay and apoptosis
pathway detection. The HepG2 cells were induced by 3.0 mmol/l VPA,
then cell apoptosis was detected by flow cytometry using an Annexin
V/propidium iodide kit. The activity of caspases 3, 8 and 9 was
detected using caspase activity detecting kits, and the activity of
the control cell was denoted as 1. Caspase protein expression was
detected by flow cytometry analysis and the mean fluorescence
intensity exponent was calculated. To detect the effect of caspase
inhibitors on the apoptosis-inducing function of VPA, the HepG2
cells were induced by a combination of 3.0 mmol/l VPA and 40 μmol/l
of the caspase 3, 8 and 9 inhibitors Z-DEVD-FMK, Z-IETD-FMK and
Z-LEHD-FMK, respectively, for 48 h, and the results were indicated
by the percent of apoptotic cells. The experiment was repeated two
times. (A) Apoptosis profile of HepG2 cells treated with VPA for
24, 48, 72 and 96 h. (B) Caspase activity inhibited by VPA.
*P<0.01 and #P>0.05. (C) Caspase
protein expression affected by VPA. *P<0.01 and
#P >0.05. (D) Effect of caspase inhibitors on HepG2
cell apoptosis. *P<0.05 and #P>0.05.
White bars, control cells; black bars, VPA-induced cells. VPA,
valproate acid sodium. |
Effect of VPA on caspase activity
Fig. 4B shows that
the activity of caspases 3 and 9 was increased by 113.0% and 86%,
respectively, by VPA, which is significantly different compared
with the control group (P<0.01). However, caspase 8 activity
exhibited no clear difference between VPA-treated and control cells
(P>0.05).
Protein expression of caspases
The protein expression of caspases in HepG2 cells
was detected by FCM analysis and was indicated by MFI. In agreement
with the caspase activity assay, the MFI of caspases 3 or 9 in
HepG2 cells treated with VPA was higher compared with untreated
cells (P<0.01), which means the protein expression of caspases 3
and 9 in HepG2 cells was markedly upregulated by VPA. However,
caspase 8 expression was only slightly affected by VPA (P>0.05;
Fig. 4C).
Caspase-blocking assay
Similar to the activity and protein expression of
the caspases, Fig. 4D shows that
the VPA-induced apoptosis of HepG2 cells could be reduced by
caspase 3 and 9 inhibitors through blocking the expression of
caspase proteins. The cell apoptosis rate was 16.9% when the cells
were induced by VPA alone. However, the apoptosis rate was only
7.6% when induced by VPA and the caspase 3 inhibitor Z-DEVD-FMK,
and was 7.2% when induced by VPA and the caspase 9 inhibitor
Z-LEHD-FMK (P<0.05). By contrast, there was no evident change in
the cell apoptosis rate when induced by VPA and the caspase 8
inhibitor Z-IETD-FMK, with a rate of 15.8% (P>0.05).
Discussion
In the 1950s, Cruft reported that histone could bind
to DNA and change the transcription activity (10). From then on, histones became a hot
research point (11). High
expression of HDACs is closely correlated with tumorigenesis and
tumor development (12). VPA has
been previously demonstrated to be a specific HDACI, but its
antineoplastic function has not been widely noticed (13).
In the present study, VPA was demonstrated to be an
HDACI, as HDAC activity and the HDAC1 gene expression of
hepatocellular carcinoma cells was inhibited by it. As a result,
cell proliferation inhibition by VPA was detected.
From the inhibition results of cell proliferation
and HDAC activity, it was observed that BEL-7402 and SMMC-7721
cells were more sensitive than HepG2 cells to VPA. Therefore, HepG2
cells were used in the subsequent experiments. If the malignant
phenotype of HepG2 cell can be reversed by VPA, the malignant
phenotype of SMMC-7721 and BEL-7402 cells should also be
reversed.
Abnormal expression of cyclin proteins is associated
with the genesis and prognosis of certain tumors (14,15).
The expression of cyclins D1, A and E significantly increases in
tumor tissue compared with normal tissue (1,16).
Genetic transcription and protein expression of cyclin A, D and E
can be blocked by P21. The P21Waf/cip1 gene is a
cyclin-dependent kinase (CDK) inhibitor that is important for
repairing DNA injury and correcting DNA replication (17). If the DNA is damaged at the
G1 phase, P21Waf/cip1 genetic transcription
is activated and binds to the cyclin and CDK, resulting in the
cyclin-CDK compound losing its kinase activity. Therefore,
cell-cycle arrest at the G1 phase and DNA replication is
inhibited, so cell growth is interrupted. It has been reported that
histone deacetylation is the key mechanism of
P21Waf/cip1 inactivation in gastric carcinoma cell lines
(18–21). The present results revealed that,
following treatment with VPA for 48 h, the cell cycle was arrested
at the G0/G1 phase and the mRNA and protein
expression of cyclin A and D1 was downregulated in HepG2 cells,
while the expression of P21Waf/cip1 was upregulated.
However, cyclin E was only marginally affected by VPA. The
VPA-induced G1-phase arrest in HepG2 cells may occur
through the following pathways, individually or synergistically.
VPA may upregulate P21Waf/cip1 mRNA and protein
expression, which binds to CDKs competitively with cyclins and
inhibits various cyclin-CDK compounds. VPA may also downregulate
cyclin D1 mRNA and protein expression, leading to decreased
activity of the cyclinD-CDK4/CDK6 pathway, so cell proliferation
does not skip the G1 phase, or VPA may downregulate
cyclin A mRNA and protein expression, followed by a reduction in
the synthesis of cyclin A-CDK2 and cyclin A-CDK1 compounds.
Therefore, DNA synthesis of S phase cells may reduce, and the cells
are prevented from switching to M phase from G2 phase
and, as a result, the cells arrested at the G1 phase. In
the majority of cases, cell cycle arrest is the directional step
prior to cell apoptosis.
In view of apoptosis as one mechanism for antitumor
proliferation, the present study focused on the function that VPA
induces apoptosis in hepatocellular carcinoma cells, and aimed to
determine the contribution of the intrinsic or extrinsic pathways
to the apoptosis. Firstly, the VPA-induced apoptosis of HepG2 cells
was identified by FCM using an Annexin V/PI apoptosis detection
kit. Caspases are the center components of apoptosis, and the
cascade reaction of caspases is the activator (22,23).
Caspase 3 is the most important effector molecule in the caspase
family and is located at the termini of the apoptosis process,
termed the apoptosis executor. Caspases 8 and 9 are promoters of
the intrinsic and extrinsic apoptosis pathway, respectively. The
present results revealed that, following treatment with VPA, the
activity and protein expression of caspases 9 and 3 was markedly
upregulated, but caspase 8 expression was not evidently changed.
Furthermore, HepG2 cell apoptosis could be reduced by inhibitors of
caspases 3 and 9, but not by caspase 8 inhibitors. All these
indicate that VPA could induce the apoptosis of HepG2 cells by
activating the intrinsic or mitochondrion pathway.
The acetylation level of the histone N terminal can
alter the condition of chromatin by interfering with the affinity
of histones for DNA, or by disturbing the combination of
transcription factors and DNA sequence (24). The regulatory role of acetylation in
gene expression is similar to the DNA genetic code, and so the role
of HDACIs was observed in tumor therapy. The present study revealed
that regulating histone acetylation with VPA can markedly reverse
the malignant phenotype of hepatoma carcinoma cells, confirming
that it can be widely applied in hepatoma treatment. However, it
was the intrinsic apoptosis pathway that contributed to the
induction of apoptosis by VPA in hepatocellular carcinoma
cells.
Acknowledgements
The authors acknowledge the financial support of the
Natural Science Foundation of Shandong Province (grant no.
ZR2009CL017).
References
|
1
|
Turner BM: Cellular memory and the histone
code. Cell. 111:285–291. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mottet D and Castronovo V: Histone
deacetylases: target enzymes for cancer therapy. Clin Exp
Metastasis. 25:183–189. 2008. View Article : Google Scholar
|
|
3
|
Weichert W, Röske A, Gekeler V, et al:
Association of patterns of class I histone deacetylase expression
with patient prognosis in gastric cancer: a retrospective analysis.
Lancet Oncol. 9:139–148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marks P, Rifkind RA, Richon VM, et al:
Histone deacetylases and cancer: causes and therapies. Nature.
1:194–202. 2001.
|
|
5
|
Mork CN, Faller DV and Spanjaard RA: A
mechanistic approach to anticancer therapy: targeting the cell
cycle with histone deacetylase inhibitors. Curr Pharm Des.
11:1091–1104. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pan LN, Lu J and Huang B: HDAC inhibitors:
a potential new category of anti-tumor agents. Cell Mol Immunol.
4:337–343. 2007.PubMed/NCBI
|
|
7
|
Gallinari P, Di Marco S, Jones P, et al:
HDACs, histone deacetylation and gene transcription: from molecular
biology to cancer therapeutics. Cell Res. 17:195–211.
2007.PubMed/NCBI
|
|
8
|
Eyal S, Yagen B, Sobol E, et al: The
activity of antiepileptic drugs as histone deacetylase inhibitors.
Epilepsia. 45:737–744. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen MS, Li JQ, Zhang YQ, et al: High-dose
iodized oil transcatheter arterial chemoembolization for patients
with large hepatocellular carcinoma. World J Gastroenterol.
8:74–78. 2002.PubMed/NCBI
|
|
10
|
Cruft HJ, Mauritzen CM and Stedman E:
Abnormal properties of histones from malignant cells. Nature.
174:580–585. 1954. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi Y, Lan F, Matson C, et al: Histone
demethylation mediated by the nuclear amine oxidase homolog LSD1.
Cell. 119:941–953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Seligson DB, Horvath S, Shi T, et al:
Global histone modification patterns predict risk of prostate
cancer recurrence. Nature. 435:1262–1266. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang H, Hoshino K, Sanchez-Gonzalez B, et
al: Antileukemia activity of the combination of
5-aza-2′-deoxycytidine with valproic acid. Leuk Res. 29:739–748.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou Q, He Q and Liang LJ: Expression of
p27, cyclin E and cyclin A in hepatocellular carcinoma and its
clinical significance. World J Gastroenterol. 9:2450–2454.
2003.PubMed/NCBI
|
|
15
|
Catalano MG, Fortunati N, Pugliese M, et
al: Valproic acid induces apoptosis and cell cycle arrest in poorly
differentiated thyroid cancer cells. J Clin Endocrinol Metab.
90:1383–1389. 2005. View Article : Google Scholar
|
|
16
|
Buolamwini JK: Cell cycle molecular
targets in novel anticancer drug discovery. Curr Pharm Des.
6:379–392. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsou MF and Stearns T: Mechanism limiting
centrosome duplication to once per cell cycle. Nature. 442:947–951.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shin JY, Kim HS, Park J, et al: Mechanism
for inactivation of the KIP family cyclin-dependent kinase
inhibitor genes in gastric cancer cells. Cancer Res. 60:262–265.
2000.PubMed/NCBI
|
|
19
|
Park HY, Kim MK, Moon SI, et al: Cell
cycle arrest and apoptotic induction in LNCaP cells by MCS-C2,
novel cyclin-dependent kinase inhibitor, through p53/p21WAF1/CIP1
pathway. Cancer Sci. 97:430–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pulukuri SM and Rao JS: Activation of
p53/p21Waf1/Cip1 pathway by 5-aza-2′-deoxycytidine inhibits cell
proliferation, induces pro-apoptotic genes and mitogen-activated
protein kinases in human prostate cancer cells. Int J Oncol.
26:863–871. 2005.PubMed/NCBI
|
|
21
|
Luong QT, O’Kelly J, Braunstein GD, et al:
Antitumor activity of suberoylanilide hydroxamic acid against
thyroid cancer cell lines in vitro and in vivo. Clin Cancer Res.
12:5570–5577. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Logue SE and Martin SJ: Caspase activation
cascades in apoptosis. Biochem Soc Trans. 36:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li J and Yuan J: Caspases in apoptosis and
beyond. Oncogene. 27:6194–6206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Esteller M: Cancer epigenomics: DNA
methylomes and histone-modification maps. Nat Rev Genet. 8:286–298.
2007. View
Article : Google Scholar : PubMed/NCBI
|