Introduction
Blastic plasmacytoid dendritic cell neoplasm (BPDCN)
is a rare and highly invasive type of malignant hematopoietic and
lymphoid tissue tumor (1). Since
BPDCN was initially reported by Adachi et al (2) in 1994, it has been successively
reported in the literature. In 2004, Chaperot et al
(3) identified that the BPDCN tumor
functions similarly to plasmacytoid dendritic cells (pDC), and,
thus, proposed that it may be derived from the precursor of the
pDC. Subsequent studies determined that the BPDCN tumor cells
express the highly specific pDC markers, blood dendritic cell
antigen (BDCA)-2/cluster of differentiation (CD)303 and
BDCA-4/CD304, supporting the hypothesis that the BPDCN tumor is
derived from pDC (4). In 2005, the
World Health Organisation (WHO) European Organization for Research
and Treatment of Cancer classification of cutaneous lymphomas
recommended the use of the term CD4+/CD56+
hematodermic neoplasm (5,6). In 2008, the tumor was officially named
BPDCN in the WHO classification of lymphoid and hematopoietic
tumors and was listed as a novel, independent type of hematopoietic
and lymphoid tissue disease (7).
BPDCN can occur in individuals of all ages (range, 8
months-103 years), however, it predominantly occurs in the elderly
(8). Skin involvement is the most
prominent clinical feature and includes isolated, confined or
generalized plaques or nodules. The plaque diameter ranges from a
few millimeters to over ten centimeters, while the color ranges
from dark red to characteristic purple, and ulcers occasionally
occur. The manifestations of this disease also occasionally involve
the mucosae (9,10). In addition to the initial
manifestation of skin lesions, the disease involves other systems.
For example, lymphadenectasis occurs in 40–50% of patients, the
bone marrow and peripheral blood are involved in 60–90% of
patients, and splenomegaly occurs in just 20% of patients (1). The central nervous system is rarely
involved during the early stages of disease onset, however, it is
frequently involved in cases of disease recurrence. Group B
symptoms, such as fever, night sweats and weight loss, seldom
occur. Auxiliary examination often identifies pancytopenia and most
commonly, thrombocytopenia. Furthermore, leukemia is a common
feature in the end stage of progressive or recurrent cases, and
10–20% of BPDCN cases are accompanied by, or can progress to, acute
myeloid leukemia (11).
Case report
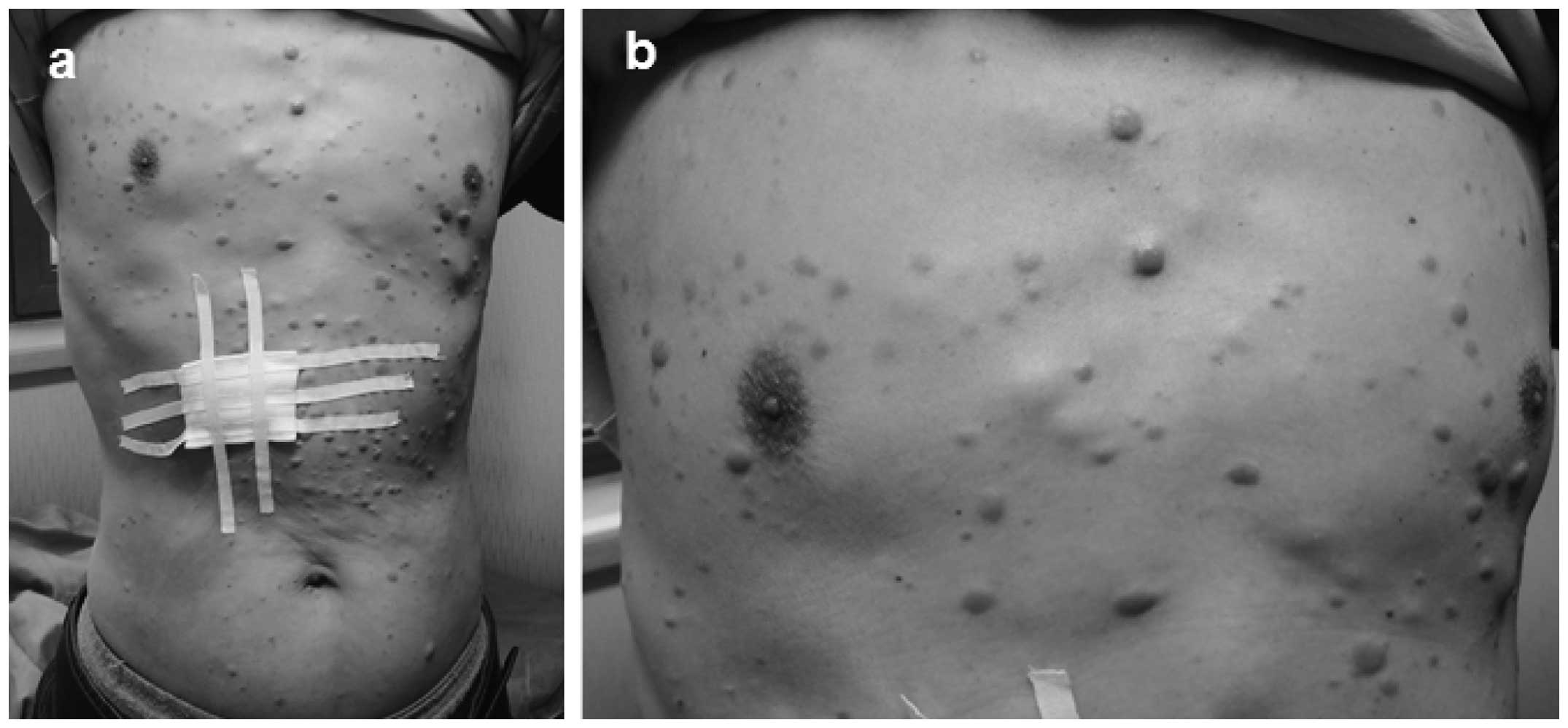
On 30th January, 2013, a 54-year-old male presented
to the Department of Dermatology, Second Affiliated Hospital of
Xi’an Jiaotong University (Xi’an, China) with systemic multiple
nodules and lumps accompanied by pain in the limbs. Physical
examination did not identify any evident abnormalities of the
heart, lungs or abdomen, however, dozens of multiple papules,
plaques and subcutaneous nodules were identified, predominantly
distributed on the trunk (Fig. 1),
with a few scattered on the head and limbs. The characteristics of
these plaques and nodules were as follows: Different textures
(hard, tough, subcutaneously located or protruding out of the
skin); different sizes (diameter range, 0.5–5.0 cm); poor motility
(with the largest on the left shoulder); varying colors (pale pink,
skin color or prunosus); smooth surface with no scales; and a
certain degree of pain caused by applying pressure. Furthermore,
the hair, mucous membranes, fingernails and toenails of the patient
appeared normal. Lymph nodes (size, 1–2 cm) on the left armpit, and
each side of the neck and groin were palpable. These nodes were
rough, hard and were not painful.
Data from a routine blood and liver kidney function
test were normal, however, a bone marrow biopsy demonstrated active
hyperplasia, 27% promyelocytic leukemia cells and positive
leukocyte peroxidase (myeloperoxidase) staining. Additionally,
pathological examination of the abdominal skin lesions identified
that the epidermis was not involved, however, the dermis and
subcutaneous fat layer were infiltrated with diffused and dense
medium-sized tumor cells (Fig. 2).
The characteristics of these tumor cells were as follows: All cells
were of a similar size and lymphoblast-like form; the chromatin was
fine and smooth; the nucleoli were prominent, with a round or oval
shape; insignificant vascular proliferation occurred, with no
vascular invasion or necrosis; and minimal inflammatory cell
infiltration was observed. Immunohistochemical staining with
leukocyte common antigen (LCA), CD4, CD56 and CD43 was positive
(Fig. 3), however, staining with
CD3, CD7, CD8, CD20, CD30, CD34, CD68 (Fig. 4) CD123, myeloperoxidase,
Epstein-Barr virus-encoded RNA (EBER) and terminal deoxynucleotidyl
transferase (TdT) and PAX-5 was negative (Fig. 5), with a Ki67 labeling index of
40–50%. These findings fulfilled the requirements for the diagnosis
of BPDCN as stage IIIE (12–14).
Relevant examinations did not demonstrate any chemotherapy
contraindications, thus, a cyclophosphamide, doxorubicin,
vincristine and prednisone (CHOP) chemotherapy program [750
mg/m2 intravenous (i.v.) cyclophosphamide, 1st day; 50
mg/m2 i.v. doxorubicin, 1st day; 1.4 mg/m2
i.v. vincristine, 1st day; 100 mg/m2 oral prednisone,
every day, 1st–5th day]was initiated. Following the first course of
chemotherapy, the nodules significantly decreased in size, however,
the patient developed fever, pharyngalgia and hoarseness. Despite
the administration of an antibiotic treatment (2.0 g ceftriaxone,
twice a day for 10 days), the patient developed sepsis, septic
shock, metabolic acidosis, respiratory acidosis, hypokalemia,
hyponatremia, hypochloremia, hypocalcemia, hypoxemia, cardiac
insufficiency, bone marrow inhibition, agranulocytosis and extreme
thrombocytopenia. The patient succumbed nine days after the first
course of chemotherapy had ended.
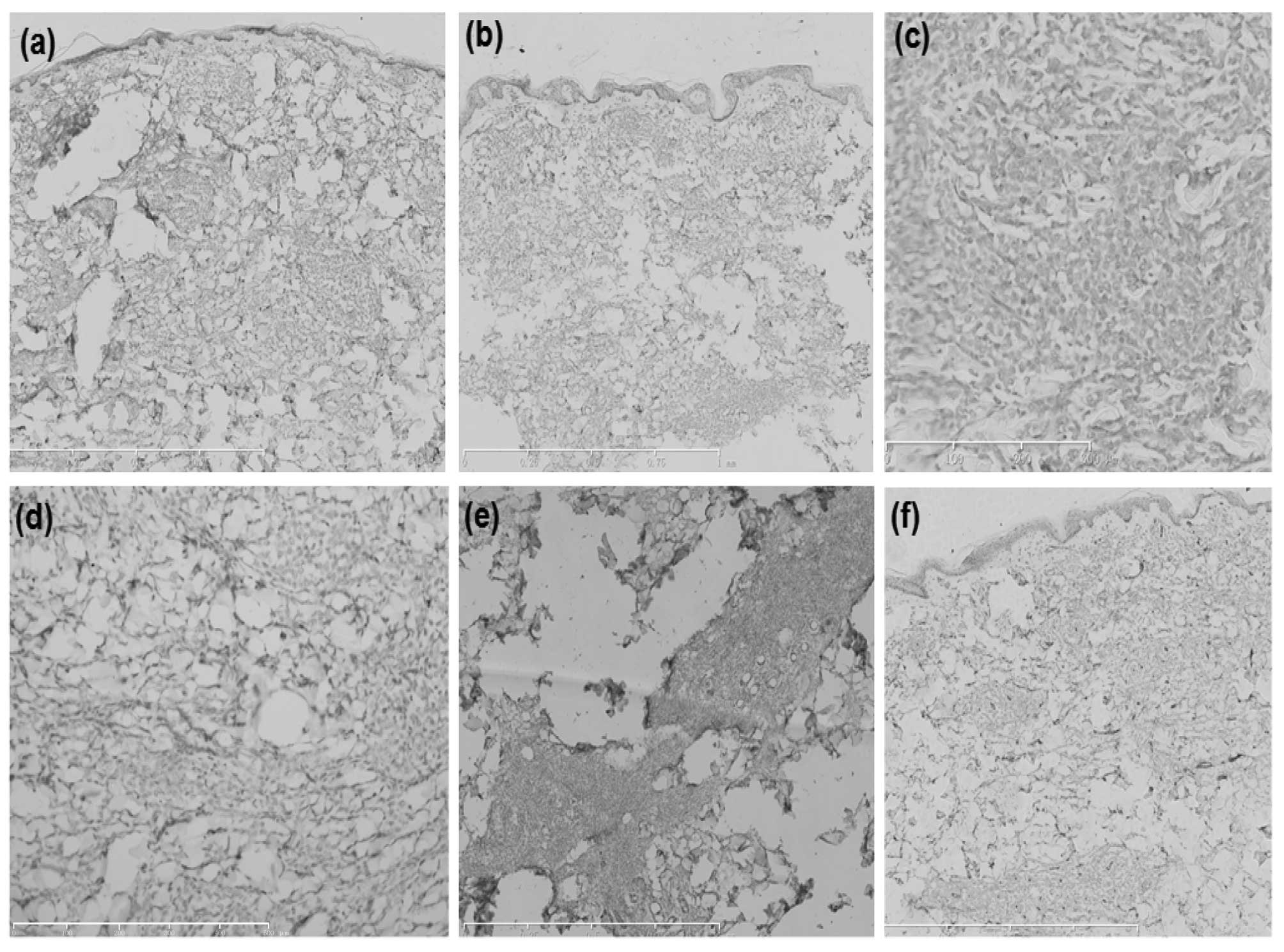 | Figure 4Immunohistochemical analysis of the
abdominal skin lesions determined that the tumor cells were
negative for (a) cluster of differentiation (CD)3 (scale bar, 1
mm), (b) CD7 (scale bar, 1 mm), (c) CD8 (scale bar, 300 μm), (d)
CD20 (scale bar, 500 μm), (e) CD30 (scale bar, 500 μm) and (f) CD34
(scale bar, 1 mm). |
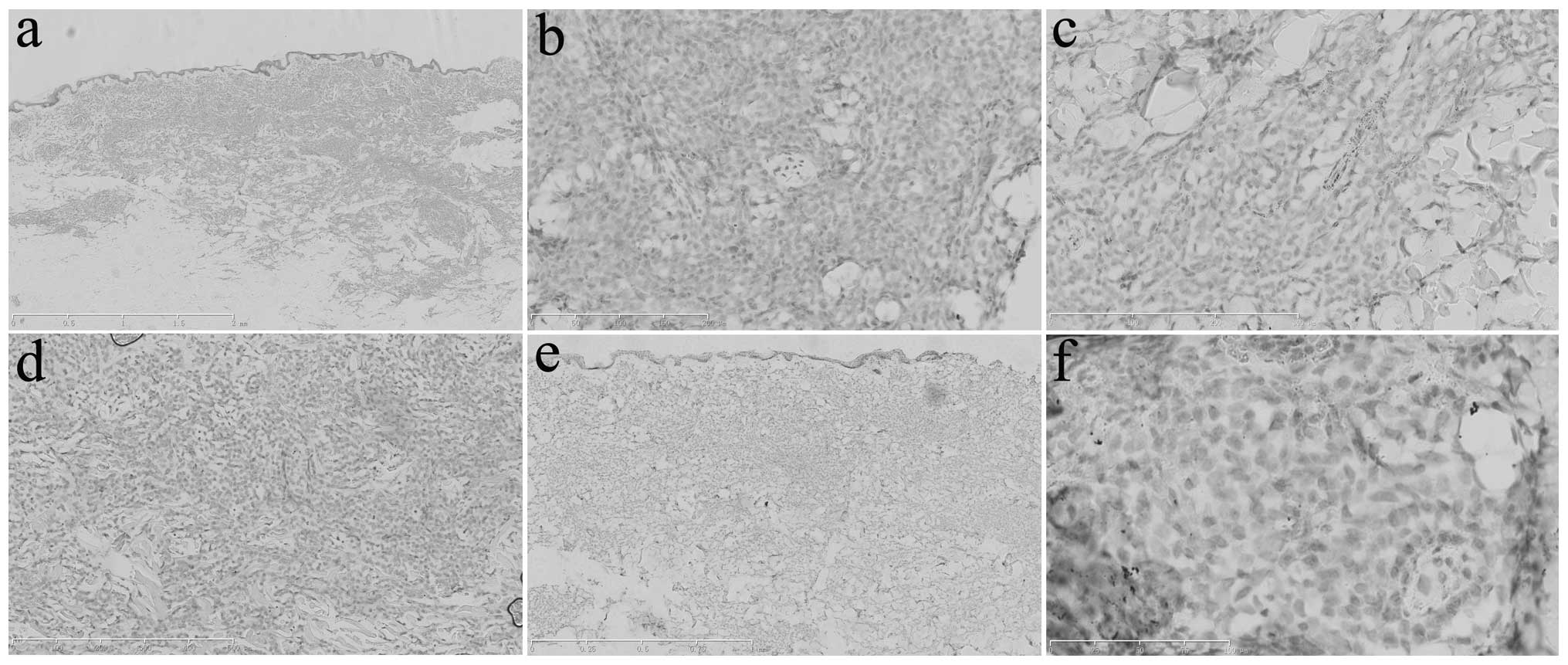 | Figure 5Immunohistochemical analysis of the
abdominal skin lesions determined that the tumor cells were
negative for (a) cluster of differentiation (CD)68 (scale bar, 2
mm), (b) CD123 (scale bar, 200 μm), (c) myeloperoxidase (scale bar,
300 μm), (d) Epstein-Barr virus-encoded RNA (scale bar, 500 μm),
(e) terminal deoxynucleotidyl transferase (scale bar, 1 mm) and (f)
PAX-5 (scale bar, 100 μm). |
Discussion
The present study describes a case of BPDCN
presenting with skin lesions. The case presented with an initial
typical manifestation of skin involvement, accompanied by lymph
node and bone marrow involvement. The skin tissue pathology was
typical of BPDCN, and immunohistochemistry demonstrated positive
LCA, CD4, CD56 and CD43 staining, and excluded a myeloid and B or T
cell lineage and origin. Furthermore, immunohistochemistry
demonstrated that the tumor was EBER-negative, indicating that the
disease was not associated with EB virus infection. Thus, the
disease was eventually diagnosed as BPDCN (stage IIIE). The unique
aspect of the present case was that the pDC-specific marker, CD123,
was negative. The patient experienced rapid disease progression.
Due to personal reasons, the patient did not undergo allogeneic
hematopoietic stem cell transplantation, instead, CHOP chemotherapy
was administered. However, this program exhibited a poor curative
effect and the patient succumbed nine days after the first course
of chemotherapy ended.
BPDCN is a rare form of lymphoma-like disease
(1). The pathogenesis of BPDCN
remains unclear. Wiesner et al (15) investigated histopathological skin
lesion specimens of 14 BPDCN patients and demonstrated that
chromosomes 9, 12, 13 and 15 were more likely to be absent, and
that the cyclin dependent kinase inhibitor (CDKN) 1B site was most
commonly not present (detectable in 64% of the tumors).
Furthermore, CDKN2A-alternative reading frame-CDKN2B sites occurred
in 50% of the cases. These results indicate that the mutation of
the key cell cycle regulatory proteins p27, p16 and retinoblastoma
1 may be important in the change of the degree of malignancy in
BPDCN (15).
BPDCN has a unique immunophenotype, as it is
CD4+ and CD56+, and is generally
CD43+. CD7 and CD2 are expressed in varying degrees, and
in approximately half of all cases, tumor cells are
CD68+ and exhibit a specific cytoplasmic granular shape
(i.e., multifocal color imaging of the Golgi apparatus). Certain
cases are TdT+ (range, ~10–80%), while S-100 protein
positivity has also been demonstrated (16). Additionally, BPDCN tumor cells often
express the pDC-specific surface marker CD123. T cell
leukemia/lymphoma 1, CD2-associated protein, BDCA-2/CD303,
BDCA-4/CD304 and other pDC-associated antigens all increase the
basis for diagnosis (17,18). CD123 is the α chain of the
interleukin-3 receptor, and is a sensitive and specific marker of
pDC (7,17,18).
As it has been demonstrated that BPDCN cells are derived from pDCs,
certain studies have proposed that CD123 may be a specific marker
for the diagnosis of BPDCN (19–22).
However, CD123 is not expressed in all BPDCNs (23) and can be highly expressed in normal
granulocytes, as well as in acute granulocytic leukemia,
histiocytosarcoma and Langerhans cell histiocytosis psychosis.
A diagnosis of BPDCN is typically determined based
on histopathological and immunohistochemical examinations. In
general, BPDCN can only be diagnosed when the tumor cells
demonstrate a blastic morphology, a
CD4+/CD56+ immunophenotype, and no myeloid
and T or B cell-specific surface marker expression. Additionally,
these immunohistochemical properties are used to distinguish
between BPDCN and other skin diseases caused by leukemia. However,
it is important to be aware of the atypical pathological
manifestations of BPDCN, such as tumor cell invasion solely around
blood vessels or appendages and the polymorphic tumor cells
(24).
BPDCN is characterized by high aggressiveness, rapid
progression and a poor prognosis, with patients exhibiting a median
survival time of 12–14 months. The survival time may be associated
with the tumor stage and the age and clinical manifestations of the
patient (25,26).
The present case was a typical case, whereby skin
involvement was the first manifestation, with lymph node and bone
marrow involvement. There is a typical manifestation of BPDCN in
histopathology, and immunohistochemical staining showed LCA, CD4,
CD56 and CD43 positivity, excluding B, T and myeloid cell lineage
and origin. EBER staining was negative, which indicated that there
was no association with EB virus infection. The final diagnosis was
BPDCN (stage IIIE) and, notably, CD123 was negative in this case,
which was considered as a unique cell marker for BPDCN. Due to
personal reasons, the patient did not receive allogeneic
hematopoitic stem cell transplantation, and instead received CHOP
chemotherapy. However, the treatment efficacy was poor and the
patient succumbed nine days after the first course of chemotherapy
had ended. In summary, BPDCN is a rare type of lymphatic and
hematopoietic tumor, the current understanding and effective
treatment of which remain to be further investigated.
References
|
1
|
Petrella T, Bagot M, Willemze R, et al:
Blastic NK-cell lymphomas (agranular CD4+CD56+ hematodermic
neoplasms): a review. Am J Clin Pathol. 123:662–675. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Adachi M, Maeda K, Takekawa M, et al: High
expression of CD56 (N-CAM) in a patient with cutaneous CD4-positive
lymphoma. Am J Hematol. 47:278–282. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chaperot L, Perrot I, Jacob MC, et al:
Leukemic plasmacytoid dendritic cells share phenotypic and
functional features with their normal counterparts. Eur J Immunol.
34:418–426. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boiocchi L, Lonardi S, Vermi W, Fisogni S
and Facchetti F: BDCA-2 (CD303): a highly specific marker for
normal and neoplastic plasmacytoid dendritic cells. Blood.
122:296–297. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burg G, Kempf W, Cozzio A, et al:
WHO/EORTC classification of cutaneous lymphomas 2005: histological
and molecular aspects. J Cutan Pathol. 32:647–674. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Willemze R, Jaffe ES, Burg G, et al:
WHO-EORTC classification for cutaneous lymphomas. Blood.
105:3768–3785. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Swerdlow SH, Campo E, Harris NL, Jaffe ES,
Pileri SA, Stein H, Thiele J and Vardiman JW: WHO Classification of
Tumours of Haematopoietic and Lymphoid Tissues. 2. 4th edition.
IARC Press; Lyon: 2008
|
|
8
|
Dalle S, Beylot-Barry M, Bagot M, et al:
Blastic plasmacytoid dendritic cell neoplasm: is transplantation
the treatment of choice? Br J Dermatol. 162:74–79. 2010. View Article : Google Scholar
|
|
9
|
Foong HB, Chong M, Taylor EM, Carlson JA
and Petrella T: Blastic plasmacytoid dendritic cell neoplasm in an
elderly woman. Med J Malaysia. 68:161–163. 2013.PubMed/NCBI
|
|
10
|
Maio P, Fernandes C, Afonso A, Sachse F,
Cabecadas J and Cardoso J: Case for diagnosis. An Bras Dermatol.
88:131–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garnache-Ottou F, Chaperot L, Biichle S,
et al: Expression of the myeloid-associated marker CD33 is not an
exclusive factor for leukemic plasmacytoid dendritic cells. Blood.
105:1256–1264. 2005. View Article : Google Scholar
|
|
12
|
Eichenauer D, Engert A, Diehl V, et al:
Hodgkin lymphoma: clinical manifestations, staging, and therapy.
Hematology: Basic Principles and Practice. Hoffman R, Benz EJ,
Leslie E and Silberstein LE: 6th edition. Elsevier; Saunder,
Philadelphia, PA: pp. 1138–1156. 2013
|
|
13
|
Kaushansky K, Lichtman MA, Beutler E, et
al: General Considerations of Lymphoma: Epidemiology, Etiology,
Heterogeneity, and Primary Extranodel Disease. William’s
Hematology. 8th edition. McGraw-Hill Medical; New York, NY: pp.
1497–1510. 2010
|
|
14
|
Macon WR, McCurley TL, Kurtin PJ and Dogan
A: Lymphoproliferative disorders. Wintrobe’s Clinical Hematology.
Wintrobe MM and Greer JP: 12th edition. Lippincott Williams &
Wilkins; Philadelphia, PA: pp. 2017–2311. 2009
|
|
15
|
Wiesner T, Obenauf AC, Cota C, Fried I,
Speicher MR and Cerroni L: Alterations of the cell-cycle inhibitors
p27(KIP1) and p16(INK4a) are frequent in blastic plasmacytoid
dendritic cell neoplasms. J Invest Dermatol. 130:1152–1157. 2010.
View Article : Google Scholar
|
|
16
|
Bilbao EA, Chirife AM, Florio D, Gimenez
LB, Marino L and Rosso DA: Hematodermic CD4+
CD56+ neoplasm in childhood. Medicina (B Aires).
68:147–150. 2008.(In Spanish).
|
|
17
|
Marafioti T, Paterson JC, Ballabio E, et
al: Novel markers of normal and neoplastic human plasmacytoid
dendritic cells. Blood. 111:3778–3792. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dzionek A, Inagaki Y, Okawa K, et al:
Plasmacytoid dendritic cells: from specific surface markers to
specific cellular functions. Hum Immunol. 63:1133–1148. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lencastre A, Cabete J, João A, Farinha P,
Ferreira G and Lestre S: Blastic plasmacytoid dendritic cell
neoplasm. An Bras Dermatol. 88(6 Suppl 1): 158–161. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shi Y and Wang E: Blastic plasmacytoid
dendritic cell neoplasm: a clinicopathologic review. Arch Pathol
Lab Med. 138:564–569. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Riaz W, Zhang L, Horna P and Sokol L:
Blastic plasmacytoid dendritic cell neoplasm: update on molecular
biology, diagnosis, and therapy. Cancer Control. 21:279–289.
2014.PubMed/NCBI
|
|
22
|
Julia F, Dalle S, Duru G, et al: Blastic
plasmacytoid dendritic cell neoplasms: clinico-immunohistochemical
correlations in a series of 91 patients. Am J Surg Pathol.
38:673–680. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cota C, Vale E, Viana I, et al: Cutaneous
manifestations of blastic plasmacytoid dendritic cell
neoplasm-morphologic and phenotypic variability in a series of 33
patients. Am J Surg Pathol. 34:75–87. 2010. View Article : Google Scholar
|
|
24
|
Kharfan-Dabaja MA, Lazarus HM, Nishihori
T, Mahfouz RA and Hamadani M: Diagnostic and therapeutic advances
in blastic plasmacytoid dendritic cell neoplasm: a focus on
hematopoietic cell transplantation. Biol Blood Marrow Transplant.
19:1006–1012. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Suzuki R, Nakamura S, Suzumiya J, et al:
Blastic natural killer cell lymphoma/leukemia (CD56-positive
blastic tumor). Cancer. 104:1022–1031. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jegalian AG, Buxbaum NP, Facchetti F, et
al: Blastic plasmacytoid dendritic cell neoplasm in the pediatric
population: diagnostic features and clinical implications.
Haematologica. 95:1873–1879. 2010. View Article : Google Scholar : PubMed/NCBI
|