Introduction
Worldwide, cervical cancer is the third most
frequently diagnosed cancer and is the fourth leading cause of
cancer-associated mortality in females, accounting for 9% of all
novel cancer cases and 8% of all cancer-associated mortalities in
females in 2008 (1). The
development of cervical cancer is associated with recurrent
high-risk human papillomavirus (HPV) infection, particularly HPV16,
which exhibits a prevalence of ~60% (2–5). The
E6 and E7 proteins of HPV16 play an important role in the
occurrence and development of HPV16-induced cervical cancer
(6). Therefore, the E6 and E7
oncogenic proteins are good targets for developing therapeutic
vaccines for cervical cancer (7,8).
Immunization with E6 or E7 long peptide-containing human leukocyte
antigen epitopes or recombinant proteins may generate potent
antitumor immunity (9–14). In previous studies, it has been
found that immunization with the HPV16 E7 protein elicited specific
protective immunity against TC-1 tumor growth (15), and mouse autologous heat shock
protein 70 (HSP70) enhanced the antitumor potency of the E7 protein
without any adjuvant (16).
It has been reported that mutations in the E6 or E7
proteins severely reduces or completely abolishes the transforming
ability of these proteins (17–19).
His-2, Cys-24 and two Cys-X-X-Cys metal-binding motifs, located at
amino acid residues 58–61 and 91–94, are the most important regions
associated with the transforming ability of the E7 protein
(19). Focal transformation of rat
3Y1 cells by E7 has been demonstrated to be eliminated by His-2 to
Asp or Cys-24 to Gly mutations and the transformation was also
considerably impaired by Cys-61 or Cys-94 to Gly mutations
(19). In order to improve the
safety of the protein vaccine, the mutation in the E7 sequence was
cloned in this study. Compared with the E7 gene, mutant
non-transforming HPV16 E7 (mE7) contains a T to G point mutation at
base 70 and removal of 15 bases from the 3′ end of the E7 protein,
which substitutes Cys-24 with Gly and disrupts the carboxyl
terminal Cys-X-X-Cys motif by deleting five amino acid residues
that contain the code for Cys-94 in the E7 protein.
Protein vaccination has become the most popular form
of HPV therapeutic vaccines as the vaccines are safe and
demonstrate no human leukocyte antigen restriction (20). In the present study, the mutant
HPV16 E7 gene (mE7) was amplified by splicing overlap extension
polymerase chain reaction (splicing PCR) using pET-28a(+)-E7 as a
template. The mE7 protein was expressed and purified in an
Escherichia coli (E. coli) system. The inhibition of TC-1
cell growth was then investigated using a TC-1 mouse model
(21). TC-1 cells derived from
primary epithelial cells of C57BL/6 mice were co-transformed with
the HPV16 E6, E7 and c-Ha-ras oncogenes. The TC-1 mouse model is a
popular model of human cervical cancer. In the present study, mice
were immunized with the mE7 protein to investigate the inhibition
of TC-1 cell growth by using this model.
Materials and methods
Materials
E. coli DH5α and Rosetta were purchased from
Invitrogen (Carlsbad, CA, USA). Yeast extract and tryptone were
obtained from Thermo Fisher Scientific (Waltham, MA, USA).
RPMI-1640 medium, fetal bovine serum, non-essential amino acids and
G418 were obtained from Gibco Life Technologies (Grand Island, NY,
USA). Plas/mini Isolation Spin-kit, complete Freund’s adjuvant
(CFA) and incomplete Freund’s adjuvant (IFA) were obtained from
Sigma-Aldrich (St. Louis, MO, USA). Sodium bicarbonate and sodium
pyruvate were obtained from Amresco LLC (Solon, OH, USA). Taq DNA
polymerase, T4 DNA ligase, restriction endonucleases and pMD18T
vector were obtained from Takara Biotechnology Co., Ltd. (Dalian,
Liaoning, China). L-glutamine was purchased from HyClone (Logan,
UT, USA). N-2-Hydroxyethylpiperazine-N′-2-ethanesulfonic acid
(HEPES) was obtained from Promega (Madison, WI, USA). Penicillin
was purchased from North China Pharmaceutical Group (Shijiazhuang,
Hebei, China), streptomycin was from Shandong Lukang Pharmaceutical
Co. (Jining, Shandong, China) and the expression vector pET-28a(+)
was from Novagen (Darmstadt, Germany). Ni-NTA agarose was obtained
from Qiagen (Valencia, CA, USA). The HPV16 E7 antibody, a mouse
monoclonal IgG1, was purchased from Santa Cruz Biotechnology (cat.
no. sc-6981; Dallas, TX, USA) and the PCR primers were synthesized
by Shanghai Sangon Biological Engineering Technology and Service
Co., Ltd. (Shanghai, China).
Cell culture
The TC-1 cell line was provided by Dr Wu of Johns
Hopkins University (Baltimore, MD, USA). The cells were maintained
in RPMI-1640, supplemented with 10% fetal bovine serum, 400 μg/ml
G418, 2 mM L-glutamine, 1.5 mg/ml sodium bicarbonate, 4.5 mg/ml
glucose, 10 mM HEPES, 1 mM sodium pyruvate, 2 mM non-essential
amino acid, 100 units/ml penicillin and 100 μg/ml streptomycin at
37°C in a 5% CO2 atmosphere.
Construction of the pET-28a(+)-mE7
expression plasmid
In a previous study, the HPV16 E7 gene was cloned
from the human cervical cancer cell line CaSKi and the prokaryotic
expression vector pET-28a(+)-E7 was constructed (15). The HPV16 mE7 gene was amplified by
splicing PCR using pET-28a(+)-E7 as a template. Two sets of primers
were used. The first set of primers was the forward primer mE7p1
that contained a site for the restriction enzyme NdeI and
the reverse primer mE7p1-G-3′, which carried a T to G mutation at
base 70 in the E7 gene (Table I).
The primers were used to amplify the sequence between the
NdeI site and base 84 (135 bp; NdeI-E784).
The second set of primers was the forward primer mE7p2-G-5′, which
carried a T to G mutation at base 70 in the E7 gene, and the
reverse primer mE7p2, which contained a site for HindIII
(Table I). The second set of
primers was used to amplify the E7 gene between bases 56 and 279,
with a UAA stop codon (236 bp; E756–279). The two PCR
products NdeI-E784 and E756–279 were
purified by electrophoresis in 2% agarose gel. mE7 was then
amplified by splicing PCR using NdeI-E784 and
E756–279 as templates. The primers were mE7p1 and
mE7p2.
 | Table IPrimer sequences of mE7 gene used for
amplification. |
Table I
Primer sequences of mE7 gene used for
amplification.
| Primer | Sequence | Restriction site |
|---|
| mE7p1 |
5′-CGCATATGGCTAGCATGACTGGTGGA-3′ | NdeI |
| mE7p1-G-3′ |
5′-TAATTGCTCATAACCGTAGAGATCAGTTG-3′ | None |
| mE7p2-G-5′ |
5′-CAACTGATCTCTACGGTTATGAGCAATT-3′ | None |
| mE7p2 |
5′-CGAAGCTTTTAGATGGGGCACACAATTC-3′ | HindIII |
PCR was then performed as previously described
(22). The conditions for PCR
amplification were as follows: 94°C for 3 min; 35 cycles of 94°C
for 45 sec, 56°C for 1 min, and 72°C for 45 sec; and a final
extension for 3 min at 72°C. The recombinant pMD18T-mE7 and
pET-28a(+)-mE7 plasmids were constructed as previously described
(15).
Expression, purification and
characterization of the mE7 protein
Expression of the mE7 protein was assessed as
previously described (15,16). Briefly, the expression of mE7 was
induced in E. coli Rosetta by isopropyl-β-D-thiogalactoside
(IPTG). The inclusion body and supernatant were analyzed by
SDS-PAGE in a 12% agarose gel. The inclusion bodies that contained
the mE7 protein were then dissolved, and the mE7 protein was
purified using Ni-NTA agarose column chromatography. The identity
and the purity of the recombinant proteins were determined by
SDS-PAGE. The concentration of the proteins was measured using a
Bradford assay (23). To confirm
the characteristics of the mE7 protein, the human HPV16 E7 antibody
was used to verify the purified protein using western blot
analysis.
Prevention and inhibition of TC-1 cell
growth by immunization with the mE7 protein
The tumor model was established using female C57BL/6
mice that were 6–8 weeks old, obtained from Shanghai Laboratory
Animal Center (Shanghai, China). All procedures performed in the
animal study were approved by the Animal Study Committee of the
Institute of Molecular Medicine (Nanjing University, Nanjing,
China).
The preventative tumor model was established
following the previously described method (15,16).
In brief, 12 mice underwent subcutaneous (SC) immunization by
either PBS or 1.5 nmol mE7 protein with CFA. A second equivalent
dose of the same solution with IFA was administered by
intraperitoneal injection two weeks later. The mice received an
additional SC injection, consisting of 1×105 TC-1 cells,
in the right flank seven days subsequent to the second
immunization. After 40 days, the tumor-free mice and a novel group
of control mice were again challenged with 2×105 TC-1
cells. The tumor growth was monitored and tumor incidence was also
recorded.
In the therapeutic tumor model, 30 mice were
administered with an SC injection consisting of 1×105
TC-1 cells in the right flank, and underwent SC immunization with
1.5 nmol mE7 protein, E7 protein or PBS in combination with CFA on
the following day. Each group contained 10 mice. One week later,
the mice were immunized by intraperitoneal injection using the same
dose of protein supplemented with IFA. Tumor growth was monitored
every three days until the control mice began to succumb. The
survival rate was recorded and the tumor volume was determined as
previously described (15).
Statistical analysis
The statistical analysis was performed as previously
described (15). All data were
expressed as the mean ± standard error of the mean. The comparison
of tumor volume between individual data points was made using a
Student’s t-test. The data for the survival percentage were
evaluated by the Log-rank test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Construction of pET-28a(+)-mE7
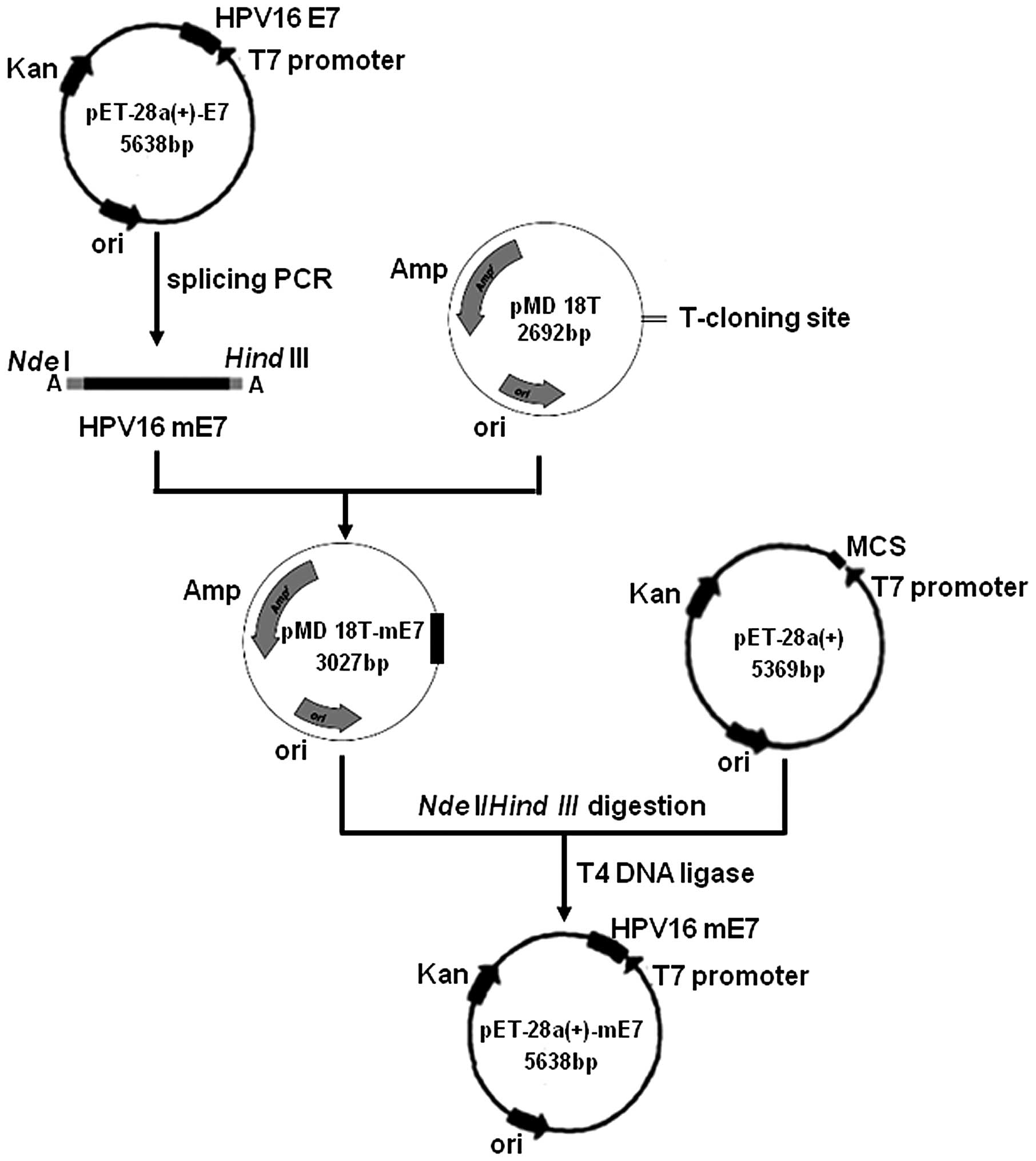
plasmid
The procedure for the construction of the
pET-28a(+)-mE7 plasmid is shown in Fig.
1. The HPV16 mE7 gene was amplified by splicing PCR using
pET-28a(+)-E7 as a template. The mE7 gene was then directly
inserted into a pMD18T vector to form the pMD18T-mE7 plasmid. The
pET-28a(+) and pMD18T-mE7 vectors were each digested with the
NdeI and HindIII restriction endonucleases.
Subsequent to purification by electrophoresis on a 2% agarose gel,
mE7 and pET-28a(+) were ligated with T4 DNA ligase to yield the
pET-28a(+)-mE7 expression plasmid. Compared with the E7 gene, mE7
was lacking nucleotides 280–294, which corresponds to residues
94–98 in the E7 protein, and nucleotide 70 in mE7 is changed from T
to G, which results in residue 24 in the E7 protein being changed
from Cys to Gly. The construction of the pET-28a(+)-mE7 plasmid
allowed the expression of the fusion mE7 protein to have a
hexa-histidine tag (his-tag) at the N-terminal end. The his-tag
sequence served as a tag for affinity purification using a Ni-NTA
agarose column.
Expression, purification and
characterization of the mE7 protein
The mE7 protein was expressed efficiently in E.
coli Rosetta subsequent to 4-h induction with IPTG, which
accounted for ~10% of total cell protein, as measured using
densitometry (Fig. 2A). The mE7
protein was expressed as an inclusion body (Fig. 2B). The mE7 protein with the his-tag
was purified using a Ni-NTA agarose column. The purification of the
mE7 protein was confirmed by SDS-PAGE (Fig. 2B). The HPV16 mE7 and E7 proteins
were each recognized by the human E7 antibody (Fig. 2C). The results of SDS-PAGE and
western blot analysis revealed that the molecular weight (MW) of
the purified mE7 was ~20 kDa, which was larger than the theoretical
MW of 14.5 kDa, and the MW of the E7 protein was 20.5 kDa, which
was also larger than the theoretical MW of 15 kDa (15). The mE7 protein was smaller than the
E7 protein as it lacked of the five amino acids at the C-terminal
end.
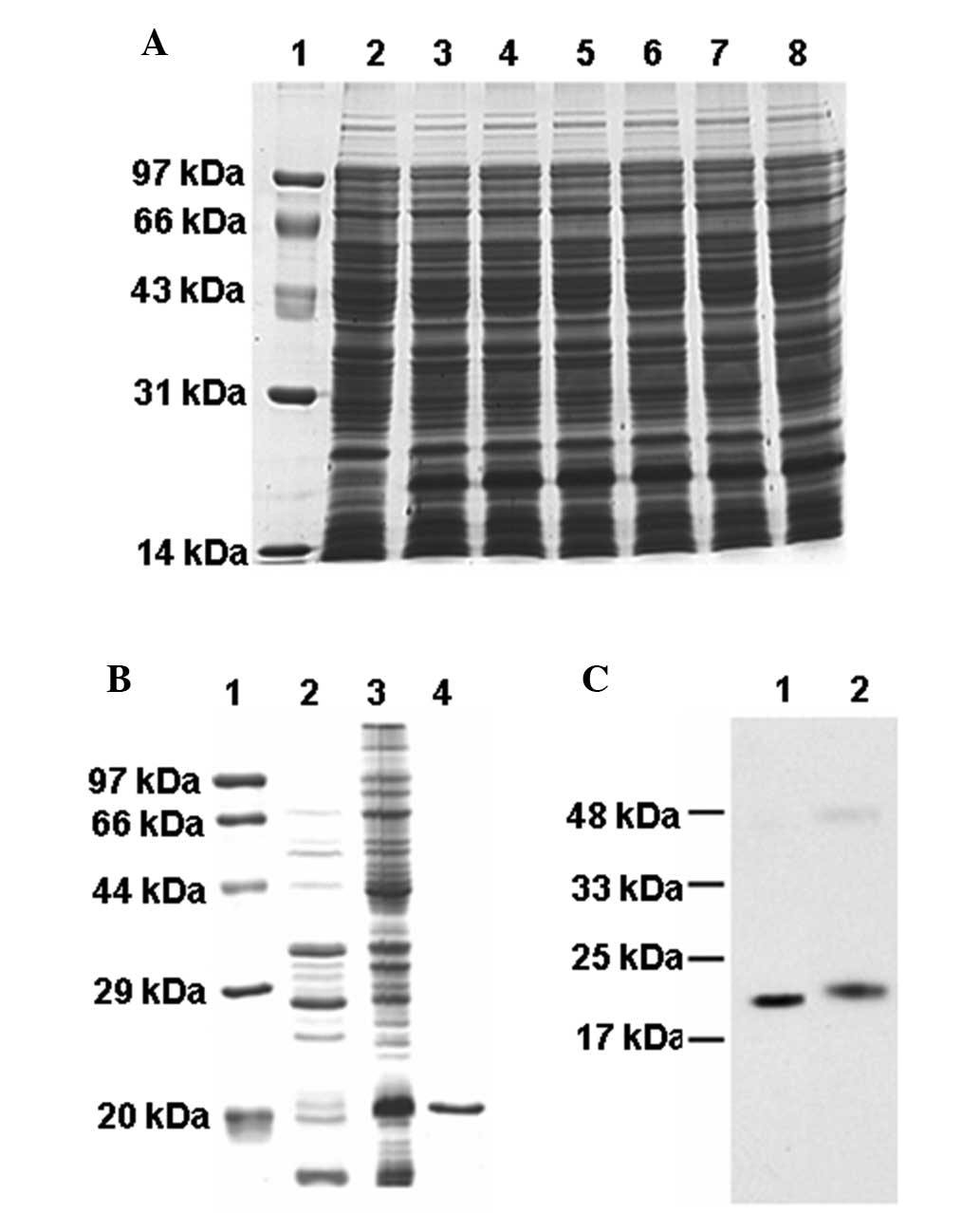 | Figure 2Expression and analysis of the mE7
protein. (A) Expression of the mE7 protein with IPTG induction.
Lane 1, protein molecular weight markers; Lane 2, total protein
extracts without IPTG induction; Lanes 3–8, total protein extracts
with IPTG induction for 1–6 h, respectively. (B) Suspension and
inclusion body of the mE7 protein following expression induction by
IPTG for 4 h. Lane 1, protein molecular weight marker; Lane 2,
suspension protein; Lane 3, inclusion body; Lane 4, purified mE7
protein. (C) Western blot analysis of (lane 1) purified mE7 and
(lane 2) E7 protein. mE7, mutant non-transforming human
papillomavirus 16 E7; IPTG, isopropyl-β-D-thiogalactoside. |
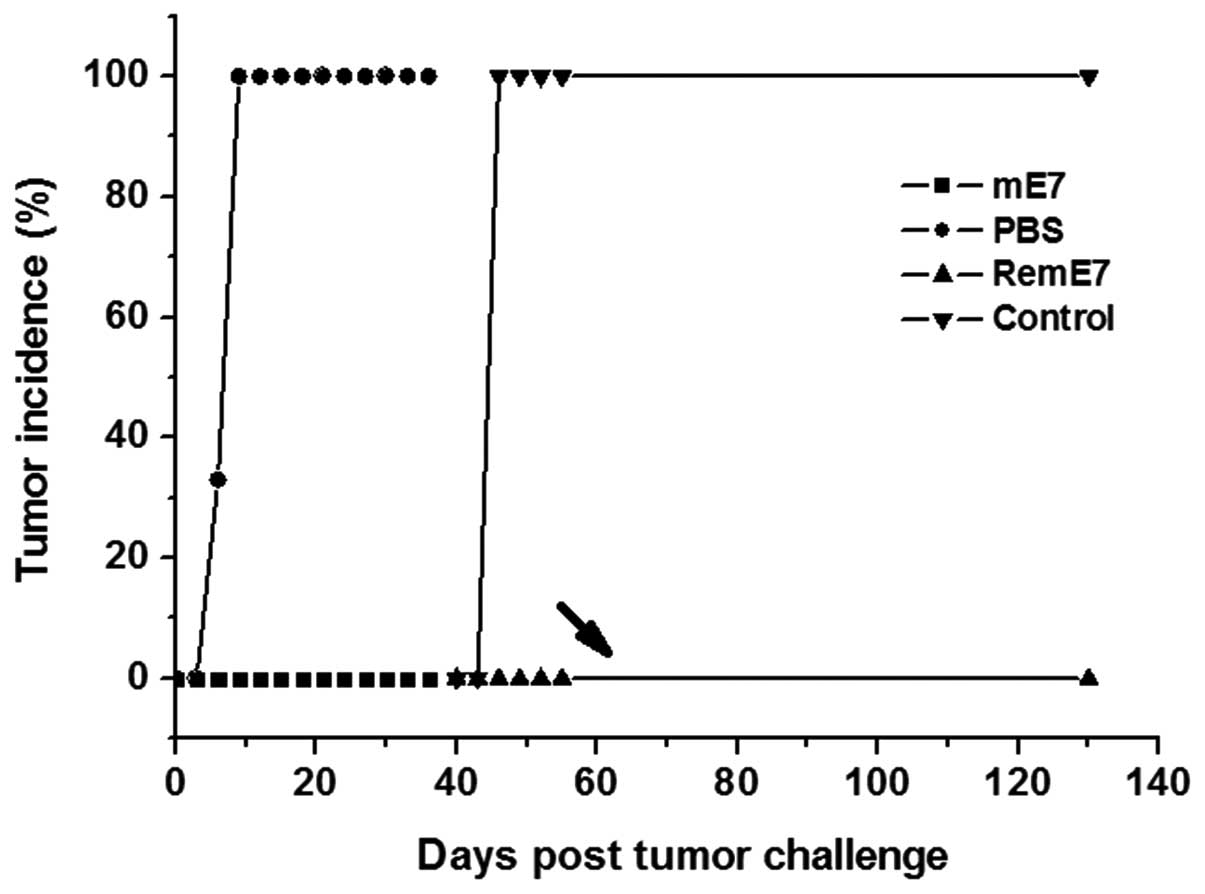
Prevention of TC-1 growth by immunization
with the mE7 protein
Female C57BL/6 mice were immunized twice with either
the mE7 protein or PBS. Subsequent to receiving an SC injection
containing 1×105 TC-1 cells, all control mice, which
were immunized with PBS, developed tumors within nine days. However
there no tumors were identified in mice immunized with the mE7
protein. After 40 days, the tumor-free mice and novel control group
mice were re-challenged with 2×105 TC-1 cells. All
control mice developed tumors within six days, but no tumor was
identified in the mE7 protein-immunized mice until 90 days
subsequent to the administration of tumor cells (Fig. 3). These results demonstrated that
immunization with the non-transforming mE7 protein was also able to
elicit a long-term protective immunity against TC-1 tumor growth
that was similar to the effect of immunization with the E7 protein
(15).
Inhibition of TC-1 cell growth by
immunization with mE7
Female C57BL/6 mice received an SC injection
consisting of 1×105 TC-1 cells in the right flank, and
were immunized with the mE7 protein, E7 protein or PBS in
combination with CFA the following day. Seven days later, the mice
were immunized using the same solution supplemented with IFA. Each
group contained 10 mice. The TC-1 cell growth was significantly
inhibited in the mice immunized with mE7 or E7compared with the
control group (P<0.05; Fig. 4A).
However, there was no significant difference in the survival rate
of the mice immunized with mE7 and the control group (P=0.055), or
E7 and the control group (P=0.105; Fig.
4B). No tumor metastasis was observed in any mouse.
Discussion
In the present study, immunization with the HPV16
mutant E7 protein was demonstrated to generate a long-term
protective immunity against TC-1 tumor growth and significantly
inhibited TC-1 tumor growth in the TC-1 mouse model.
Two HPV16 oncogenic proteins, E6 and E7, are
co-expressed in the majority of HPV16-induced cervical cancer cells
and therefore present good targets for therapeutic vaccines of
cervical cancer (4,6,8). Our
previous study reported that immunization with the full-length
HPV16 E7 protein may elicit stronger immunological protection
compared with the E6 protein, and results in a long-term specific
protective immunity against TC-1 tumor growth (15). It has been reported that the
transforming ability and trans-activation of the E7 protein was
eliminated by the Cys-24 to Gly substitution (19). The disruption of the Cys-X-X-Cys
motif that lies towards the carboxyl terminus of E7 protein
appeared to result in a greatly impaired transforming ability and
reduced transactivation (19). In
the present study, the prokaryotic expression system for the mE7
protein, the pET-28a(+)-mE7 plasmid, was constructed in order to
abolish the transforming ability of the E7 protein. Compared to the
E7 protein, the mE7 protein was lacking residues 94–98 of the E7
protein, with Cys-94 being an amino acid residue in the carboxyl
terminal Cys-X-X-Cys motif of the E7 protein, and residue 24 of the
E7 protein was changed from Cys to Gly. Therefore, vaccination with
the mE7 protein is safer compared with vaccination using the E7
protein.
The mE7 protein was further purified using a Ni-NTA
agarose column, which was confirmed by SDS-PAGE and western blot
analysis. The present results revealed that the MW of the mE7
protein was ~20 kDa, which is larger than the theoretical MW of
14.5 kDa, and the MW of the E7 protein was 20.5 kDa, also larger
than the theoretical MW of 15 kDa (15). The cause of MW variation was the
electrophoretic migration of the acidic E7 protein being altered by
the positively-charged basal amino acids of the his-tag (24). The electrophoretic migration of the
mE7 protein may possess the same property.
In order to determine the immunological effects of
the HPV16 E6 or E7-associated vaccine, the TC-1 cell line was
constructed by Dr Wu of Johns Hopkins University (21). In the present study, the TC-1
cervical cancer mice model was used to determine the effects of
vaccination with the mE7 protein using CFA or IFA, and PBS was used
as a control. To the best of our knowledge, the present results
revealed for the first time that immunization with non-transforming
mE7 protein may elicit the same protective and therapeutic immunity
against TC-1 cell growth as the E7 protein in the TC-1 cervical
cancer mice model. However, an adjuvant was required to facilitate
the anti-tumor function of the mE7 protein. HSP has been reported
to possess the potential to act as both antigen vector and adjuvant
in enhancing antigen-specific tumor immunity (25,26).
In a previous study, it was demonstrated that mouse autologous
HSP70 enhanced the antitumor potency of the E7 protein without any
adjuvant (16). The present results
demonstrated that immunization with the HPV16 mE7 protein was able
to elicit a long term protective immunity against TC-1 tumor growth
and a significant inhibition of TC-1 tumor growth in the TC-1 mouse
model. Therefore, the non-transforming mE7 and mouse autologous
HSP70 recombinant proteins are hypothesized to also be able to
elicit effective and safer anti-tumor activity compared with the
HSP70-E7 recombinant proteins.
Acknowledgements
The authors would like to thank Dr Wu of Johns
Hopkins University for providing the TC-1 cells. This study was
supported in part by a research award from Suzhou iCAT, LLC and a
grant from Shanghai Science and Technology Commission (grant no.,
11ZR141220).
References
|
1
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smith JS, Lindsay L, Hoots B, et al: Human
papillomavirus type distribution in invasive cervical cancer and
high-grade cervical lesions: a meta-analysis update. Int J Cancer.
121:621–632. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schiffman M, Castle PE, Jeronimo J,
Rodriguez AC and Wacholder S: Human papillomavirus and cervical
cancer. Lancet. 370:890–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bosch FX, Manos MM, Muñoz N, et al:
Prevalence of human papillomavirus in cervical cancer: a worldwide
perspective. International biological study on cervical cancer
(IBSCC) Study Group. J Natl Cancer Inst. 87:796–802. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Walboomers JM, Jacobs MV, Manos MM, et al:
Human papillomavirus is a necessary cause of invasive cervical
cancer worldwide. J Pathol. 189:12–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DeFilippis RA, Goodwin EC, Wu L and DiMaio
D: Endogenous human papillomavirus E6 and E7 proteins
differentially regulate proliferation, senescence, and apoptosis in
HeLa cervical carcinoma cells. J Virol. 77:1551–1563. 2003.
View Article : Google Scholar :
|
|
7
|
Accardi L, Paolini F, Mandarino A, et al:
In vivo antitumor effect of an intracellular single-chain antibody
fragment against the E7 oncoprotein of human papillomavirus 16. Int
J Cancer. 134:2742–2747. 2014. View Article : Google Scholar
|
|
8
|
Govan VA: Strategies for human
papillomavirus therapeutic vaccines and other therapies based on
the E6 and E7 oncogenes. Ann N Y Acad Sci. 1056:328–343. 2005.
View Article : Google Scholar
|
|
9
|
Ressing ME, Sette A, Brandt RM, et al:
Human CTL epitopes encoded by human papillomavirus type 16 E6 and
E7 identified through in vivo and in vitro immunogenicity studies
of HLA-A*0201-binding peptides. J Immunol. 154:5934–5943.
1995.PubMed/NCBI
|
|
10
|
Zwaveling S, Ferreira Mota SC, Nouta J, et
al: Established human papillomavirus type 16-expressing tumors are
effectively eradicated following vaccination with long peptides. J
Immunol. 169:350–358. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qian X, Lu Y, Liu Q, et al: Prophylactic,
therapeutic and anti-metastatic effects of an
HPV-16mE6Delta/mE7/TBhsp70Delta fusion protein vaccine in an animal
model. Immunol Lett. 102:191–201. 2006. View Article : Google Scholar
|
|
12
|
Liu B, Ye D, Song X, et al: A novel
therapeutic fusion protein vaccine by two different families of
heat shock proteins linked with HPV16 E7 generates potent antitumor
immunity and antiangiogenesis. Vaccine. 26:1387–1396. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou CM, Zhang GX and Ma XX:
Characterization and evaluation of the immune responses elicited by
a novel human papillomavirus (HPV) therapeutic vaccine: HPV
16E7-HBcAg-Hsp65 fusion protein. J Virol Methods. 197:1–6. 2014.
View Article : Google Scholar
|
|
14
|
Zong J, Wang C, Liu B, et al: Human hsp70
and HPV16 oE7 fusion protein vaccine induces an effective antitumor
efficacy. Oncol Rep. 30:407–412. 2013.PubMed/NCBI
|
|
15
|
Li YL, Qiu XH, Shen C, Liu JN and Zhang J:
Vaccination of full-length HPV16 E6 or E7 protein inhibits the
growth of HPV16 associated tumors. Oncol Rep. 24:1323–1329.
2010.PubMed/NCBI
|
|
16
|
Li YL, Liu J, Liu JN and Zhang J:
Immunization of protein HPV16 E7 in fusion with mouse HSP70
inhibits the growth of TC-1 cells in tumor bearing mice. Vaccine.
29:5959–5962. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dalal S, Gao Q, Androphy EJ and Band V:
Mutational analysis of human papillomavirus type 16 E6 demonstrates
that p53 degradation is necessary for immortalization of mammary
epithelial cells. J Virol. 70:683–688. 1996.PubMed/NCBI
|
|
18
|
Edmonds C and Vousden KH: A point
mutational analysis of human papillomavirus type 16 E7 protein. J
Virol. 63:2650–2656. 1989.PubMed/NCBI
|
|
19
|
Watanabe S, Kanda T, Sato H, Furuno A and
Yoshiike K: Mutational analysis of human papillomavirus type 16 E7
functions. J Virol. 64:207–214. 1990.PubMed/NCBI
|
|
20
|
Mahdavi A and Monk BJ: Vaccines against
human papillomavirus and cervical cancer: promises and challenges.
Oncologist. 10:528–538. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ji H, Chang EY, Lin KY, et al:
Antigen-specific immunotherapy for murine lung metastatic tumors
expressing human papillomavirus type 16 E7 oncoprotein. Int J
Cancer. 78:41–45. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun Z, Lu W, Tang Y, et al: Expression,
purification and characterization of human urodilatin in E. coli.
Protein Expr Purif. 55:312–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang WH ZJ, Wang ZY and Hong MM: SDS-PAGE
determination of HIS-TAG fusion protein molecular weight of the
reasons for bias. J Plant Physiol Mol Biol. 26:64–68. 2000.
|
|
25
|
Tamura Y, Peng P, Liu K, Daou M and
Srivastava PK: Immunotherapy of tumors with autologous
tumor-derived heat shock protein preparations. Science.
278:117–120. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang XY, Kazim L, Repasky EA and Subjeck
JR: Characterization of heat shock protein 110 and
glucose-regulated protein 170 as cancer vaccines and the effect of
fever-range hyperthermia on vaccine activity. J Immunol.
166:490–497. 2001. View Article : Google Scholar
|