Introduction
3H11 is a monoclonal antibody (mAb) derived from
mice sequentially immunized with varying cancer cell lines
(1). Immunohistochemical analysis
has revealed that 3H11 mAb is able to bind to various tissues from
liver, lung, bladder, breast, colon and gastric cancer (1). In addition, isotope-labeled 3H11 mAb
has been used as a radioimaging reagent in clinical tumor imaging
(2). The antigen recognized by 3H11
(3H11-Ag) has been previously cloned through cDNA library screening
(3). A bioinformatics analysis has
revealed that this novel gene (GenBank accession no., AF317887;
located on chromosome 12q21) has 17 exons and encodes 589 amino
acids. Furthermore, using northern blot analysis, a 2.3-kb
transcript has been detected, which is widely expressed in
embryonic and cancerous tissues, but not in adjacent normal tissues
(3), indicating that its function
may be associated with cancer development and progression. Based on
its expression profile and theoretical molecular weight, we defined
3H11-Ag as cancer and embryo expression protein 65 (CEP65)
(4).
Previous studies have demonstrated the presence of
high homology between CEP65 and centrosomal protein nephrocystin-6
(NPHP6), which is a protein frequently mutated in several
hereditary diseases (5). In
addition, a previous study has investigated the nuclear
distribution and revealed the potential centrosomal localization of
CEP65 (6); thus, CEP65 represents a
novel member of the centrosome-binding protein family. The
centrosome functions as a centre for microtubule organization and
is involved in cell growth, division, migration and polarity
(7). Since deregulated expression
of centrosomal proteins is closely associated with hereditary
diseases and tumor development (5,7), CEP65
may be involved in the control of malignant phenotypes of cancer
cells. The present study aimed to investigate whether CEP65
promotes cancer cell growth and invasiveness in vitro and
in vivo. Furthermore, the effect of CEP65 on matrix
metalloproteinase (MMP)2 activity and the expression levels of the
metastasis-associated genes, RAP, vitronectin (VTN)
and tissue inhibitor of metalloproteinases-2 (TIMP-2), were
investigated to establish whether CEP65 is an oncogene and a
potential target for cancer therapy.
Materials and methods
Cell culture
The human gastric cancer AGS cell line (American
Type Culture Collection, Manassas, VA, USA) was cultured in Ham’s
F-12 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with
10% fetal bovine serum (FBS; Invitrogen Life Technologies,
Carlsbad, CA, USA) in a humidified incubator containing 5%
CO2 at 37°C. Human embryonic kidney 293 (HEK-293) and
NIH3T3 cells were obtained from the American Type Culture
Collection and cultured in Dulbecco’s modified Eagle’s medium
(Bioroc Pharmaceutical & Biotech Co. Ltd., Tianjin, China)
supplemented with 10% FBS. In addition, BICR-H1 breast cancer cells
were provided by Dr Zhiqian Zhang (Peking University Cancer
Hospital and Institute, Beijing, China) and cultured in RPMI 1640
medium (Invitrogen Life Technologies) supplemented with 10%
FBS.
Generation of stable cell lines
expressing CEP65
AGS-CEP65 and AGS mock-transfected (AGS-M) cells
were generated by transfecting pcDNA3-CEP65 and pcDNA3 alone,
respectively, into AGS cells using Lipofectamine® 2000
(Invitrogen Life Technologies). The transfected cells were
subsequently selected for using 600 μg/ml G418 (Sigma-Aldrich).
After three weeks, pooled colonies were recovered. Recombinant
adenoviruses were generated using the AdEasy Adenoviral Vector
system (Agilent Technologies, Santa Clara, CA, USA), as previously
described (8). Briefly, full-length
CEP65 cDNA and antisense-CEP65 cDNA were cloned into the AdEasy
Shuttle vector, pAdTrack-cytomegalovirus. The resultant plasmids
were linearized and co-transformed into E. coli BJ5183 cells
(American Type Culture Collection) with an adenoviral backbone
plasmid, pAdEasy-1. Subsequently, the recombinants were selected
for with kanamycin. The linearized plasmids were transfected into
HEK-293 cells using Lipofectamine® 2000 to yield the
pAd-CEP65(+) and pAd-CEP65(−) viruses. The cells were infected
using a multiplicity of infection (MOI) value of 10. The expression
of exogenous CEP65 was detected using reverse transcription
polymerase chain reaction (RT-PCR) and western blot analysis.
Cell proliferation assay
In total, 100 μl of sample (2×104 cells)
was seeded in 96-well plates, using three replicates for each cell
line. The cells were harvested at 12, 24, 36, 48 and 60 h after
seeding, and the proliferation rate was measured using an MTT assay
kit from Promega Corporation (Madison, WI, USA). The absorbance
[optical density (OD) values] was measured at 492 nm using a
microplate reader (iMark; Bio-Rad Laboratories, Inc., Richmond, CA,
USA).
Cell adhesion assay
To investigate cell adhesion, 96-well microplates
were coated with 6 μg Matrigel/well (Collaborative Biomedical
Products, Atlanta, GA, USA) and allowed to dry in a laminar flow
cabinet overnight at room temperature. Subsequent to being washed
three times with PBS, the wells were blocked with 30 μl solution,
containing 20 mg/l bovine serum albumin (Sigma-Aldrich) in F-12
medium, for 1 h at 37°C. Subsequently, 8×104 cells (100
μl; in F-12 medium) were seeded into the blocked wells and allowed
to adhere for 1 h at 37°C. Following incubation, the wells were
washed three times with PBS and the number of remaining cells was
determined by an MTT assay.
Cell migration and invasion assays
A cell migration assay was performed using tissue
culture-treated 6.5-mm Transwell chambers with 8.0-μm pore
membranes, which were obtained from Corning Incorporated (Corning,
NY, USA). The bottom surfaces of the membranes were coated with 20%
FBS in F12 medium by incubation for 2 h at 37°C. Conditioned medium
was collected from a NIH3T3 cell culture and added to the bottom
chambers (800 μl/chamber) along with 50 μg/ml fibronectin. Next,
2.5×104 single AGS cells in serum-free F-12 medium (500
μl) were added to the top chamber of each Transwell. The cells were
allowed to migrate for 12 h at 37°C in an atmosphere containing 5%
CO2. Following removal of the remaining cells from the
top chamber, a cotton swab was used to clean the top surface of
each membrane. Subsequently, the cells penetrating through to the
bottom surface of the membrane were fixed in methanol. The filters
were stained in hematoxylin for 10 min, and the cells on the lower
surface of the filter were counted in five randomly selected fields
under a light microscope (Eclipse 80i; Nikon Corporation, Tokyo,
Japan) (magnification, ×200). The invasion assay was performed
using a procedure similar to the aforementioned migration assay
procedure, with the exception that the upper surface of the
membrane was coated with Matrigel and the cells were allowed to
invade for 16 h at 37°C.
In vivo cancer cell growth and metastasis
assays
All animal experiments involving SCID mice were
approved by the Medical Ethics Committee of the Peking University
Cancer Hospital and Institute, and all procedures were performed in
accordance with the animal welfare guidelines of the National
Insitute of Health (NIH Publication No. 85-23; revised 1985). To
investigate metastasis, chick chorioallantoic membrane (CAM) and
severe combined immunodeficiency (SCID) mouse model assays were
conducted as previously described (9), with minor modifications. Briefly, for
the CAM assay, BICR-H1 cells were infected with pAd, pAd-Cep65(+)
or pAd-Cep65(−). After 24 h of infection, the cells were harvested
and added onto the CAM of 10-day-old chicken embryos of the white
leghorn chicken (Gallus gallus domesticus L.; obtained from
China Agricultural University, Beijing, China). A window was cut in
the egg shell, which was sealed with Durapore tape following
addition of the cancer cells onto the CAM. The eggs were incubated
at 37.8°C and 80% relative humidity. After seven days, the tumor
weight was measured and the lungs of the chicken embryos were
checked using a fluorescence microscope (TiU; Nikon Corporation).
Embyos were sacrificed using 30 min of 90% CO2
treatment. The cells in each sample were quantified from 20 random
visual fields (x200) under a fluorescence microscope.
For the SCID mice model assay, infected BICR-H1
cells (4×106 for each) were injected into the mammary
fat pad of five-week-old female SCID mice (Weitong Lihua
Experimental Animal Technology Co., Ltd., Beijing, China). After
eight weeks, the mice were sacrificed by euthanasia and the tumor
weight was measured. The lungs of the SCID mice were removed and
analyzed by hematoxylin and eosin staining on fresh-frozen
sections.
Zymographic assay
A zymographic assay was performed according to the
procedure described by Wang et al (10), with minor modifications. Briefly,
conditioned F-12 medium was obtained from 24-h serum-deprived cells
at 70–80% confluence and concentrated by 20-fold. Concentrated
medium of 2×105 cells was electrophoresed using 10%
SDS-PAGE containing 0.1% gelatin, and incubated for 24 h in freshly
prepared developing buffer [50 mM Tris-HCl (pH 8.0), 50 mM
CaCl2 and 0.02% NaN3], following removal of
the SDS. The degree of digestion, which was assessed by the density
of the bands following staining and destaining, was proportional to
the enzymatic activity.
Western blot analysis
Cells were harvested in lysis buffer, containing 50
mM Tris-HCl (pH 7.4), 250 mM NaCl, 2 mM EDTA, 1% SDS, 2 mM
dithiothreitol and 1X protease inhibitor cocktail (Roche, Mannheim,
Germany), then sonicated using a Vibracell VCX130 Ultrasonic Cell
Disrupter (Sonics & Materials Inc., Newtown, CT, USA) for 30
sec on ice. Protein concentration was determined using a
Bicinchoninic Acid Protein Assay kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA). Cell lysates (50 μg per sample) were separated
by 10% SDS-PAGE and electrotransferred to nitrocellulose membranes.
After blocking with 5% non-fat milk in 0.1% Tris-Buffered Saline
with Tween 20, the membranes were incubated with primary monoclonal
mouse anti-human CEP65 antibody (dilution, 1:500) overnight at 4°C,
then reprobed with horseradish peroxidase-labeled goat anti-mouse
secondary antibody (dilution 1:5,000; ZSGB-BIO, Beijing, China) for
45 min at room temperature. Signals were detected by an enhanced
chemoluminescence system (Pierce Biotechnology, Inc.). Mouse
monoclonal anti-human actin antibody (1:5,000; Sigma-Aldrich) was
used to normalize loading.
cDNA microarray analysis
Total RNA was isolated from the cells using TRIzol
(Invitrogen Life Technologies). Poly (A)+ RNA was
annealed to oligo(dT) and transcribed with avian myeloblastosis
virus reverse transcriptase in the presence of Cy3-dUTP or
Cy5-dUTP. Labeled cDNAs were co-hybridized (AGS-CEP65 vs. AGS; or
AGS-CEP65 vs. AGS-MOCK) to a BiostarH-141s chip (Biostar Genechip
Inc., Shanghai, China) containing cDNAs from >14,000 human
genes. The washed slides were scanned using a ScanArray 4000 Array
scanner (PerkinElmer, Inc., Waltham, MA, USA). Following background
subtraction, filtering of inappropriate spots (including spots with
an improper morphology and intensity/background ratio, and a small
size) and global normalization, the signals were analyzed using
GenePix Pro 3.0 software (Molecular Devices, LLC, Sunnyvale, CA,
USA). Genes were considered to be downregulated or upregulated if
the difference in the ratio was >2-fold.
Semiquantitative RT-PCR
Total RNA (10 μg) was treated with DNase I (Pierce
Biotechnology, Inc.) and reverse transcribed using a reverse
transcription Kit (GoScript Reverse Transcription System, Promega
Corporation). The genes of interest were PCR-amplified using a
variable number of cycles (25–32 cycles) and 100 ng cDNA. The PCR
conditions for each cycle were as follows: Denaturation at 95°C for
30 sec, annealing at 57°C for 30 sec and extension at 72°C for 60
sec. Glyceraldehyde 3-phosphate (GAPDH) was used as an
internal control. The primers were as follows: CEP65
forward, 5′-CCCTTTCTCAAC AGACTCATATGAA-3′ and reverse,
5′CAAGGCCCACACGCTCTC-3′; MMP2 forward, 5′-TGCAATACCTGAACACCTTC-3′
and reverse, 5′-AAGGTCCATAGCTCATCGTC-3′; MMP9 forward,
5′-TCCTACTCTGCCTGCACCAC-3′ and reverse, 5′-ACAGGTCGAGTACTCCTTAC-3′;
VTN forward, 5′-TCTGCTCTTACTACCCAGAGC-3′ and reverse,
5′-GACATCTCGGATGAGCTTGG-3′; RAP forward,
5′-TTAGGATCCATGGCGCCGCGGAGGGTCAG-3′ and reverse,
5′-AGGGAATTCAGAGTTCGTTGTGCCGAGC-3′; TIMP-2 forward,
5′-TTTGCAATGCAGATGTAGTG-3′, and reverse
5′-TGGGTCCTCGATGTCGAGAAAC-3′; and GAPDH forward,
5′-ACCACAGTCCATGCCATCAC-3′ and reverse, 5′-TCCACCACCCTGTTGCTGTA-3′.
The PCR products were visualized on 1.2% agarose gel containing
0.05 μg/ml ethidium bromide.
Statistical analysis
The values are expressed as the mean ± standard
deviation from at least three independent experiments in triplicate
wells. SPSS 11.0 software (SPSS Inc., Chicago, IL, USA) was used to
perform an analysis of variance and unpaired Student’s t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
CEP65 promotes AGS cell growth and
invasiveness in vitro
Although CEP65 overexpression has been reported in
various cancerous tissue types (1),
the endogenous levels of CEP65 in >30 cancer cell lines were
undetectable on western blot analysis (data not shown). To overcome
this limitation and examine the biological effects of CEP65 in
cancer cells, the human full-length CEP65 was cloned (3) and then stably expressed in the AGS
gastric cancer cell line (AGS-CEP65), using cells expressing the
vector (AGS-M) and parental cells as controls. Western blot and
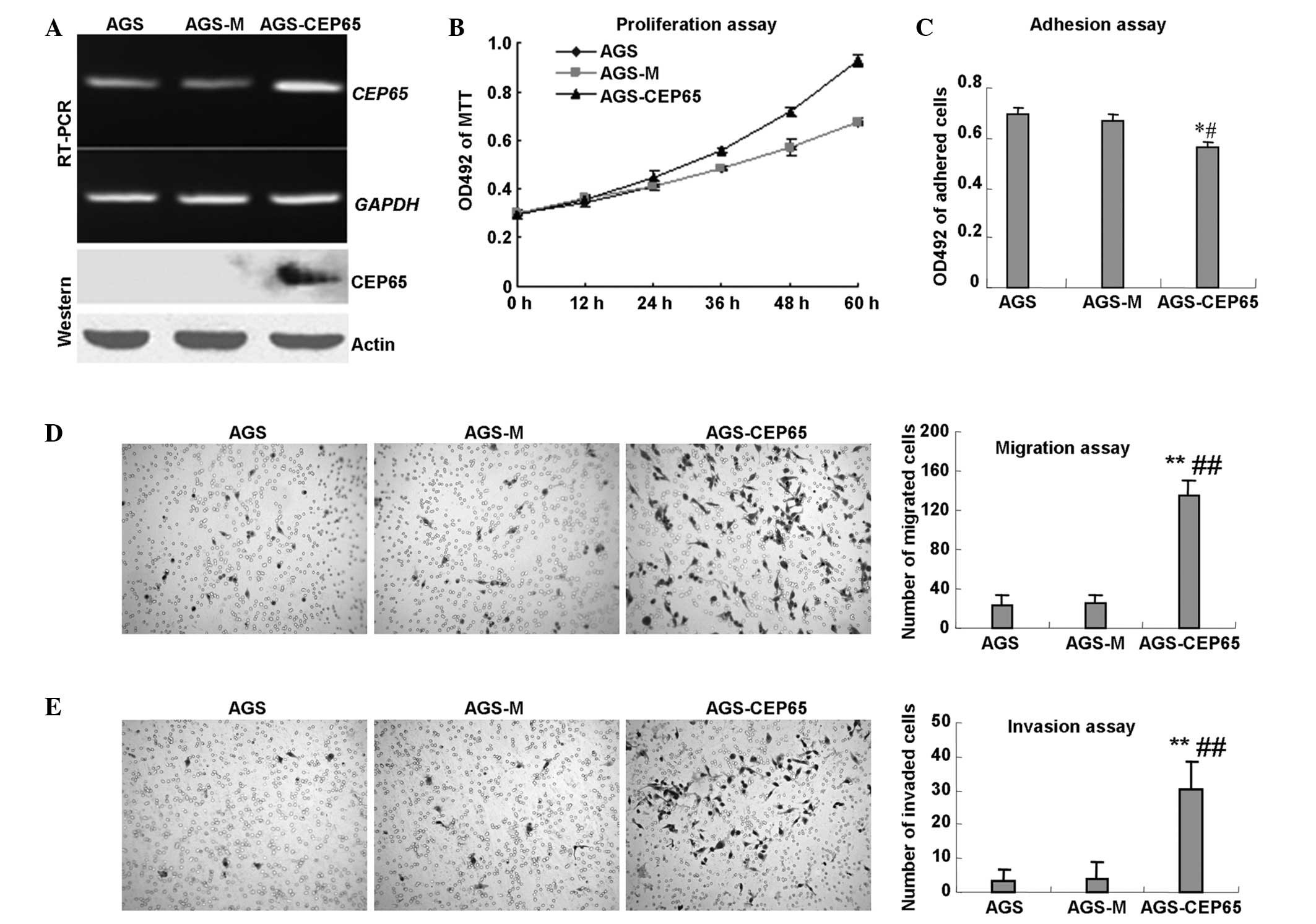
RT-PCR assays verified the expression of exogenous CEP65 (Fig. 1A). In addition, an MTT assay was
performed and CEP65 was found to significantly promote the growth
of AGS cells (Fig. 1B).
Furthermore, the adhesion ability of the AGS cells to Matrigel
mimicking extracellular matrix (ECM) was found to be reduced by
16.2 and 18.8% in the AGS-CEP65 cells compared with the AGS-M and
AGS cells, respectively (P<0.05; Fig. 1C). Next, cell migration and invasion
assays were performed to investigate the invasiveness of the
AGS-CEP65 cells. As shown in Fig.
1D, the motility was increased by 4.1- and 4.7-fold in the
AGS-CEP65 cells compared with the AGS-M and AGS cells, respectively
(P<0.01). The results of the invasion assay (Fig. 1E) revealed that the invasive
capacity was also increased by 6.9- and 8.4-fold in the AGS-CEP65
cells compared with the AGS-M and AGS cells, respectively
(P<0.01). No statistically significant differences were observed
between AGS-M and AGS in the migration or invasion assays
(P>0.05). These results indicated that CEP65 promoted cancer
cell growth and invasiveness in vitro.
CEP65 promotes BICR-H1 cell growth and
metastasis in vivo
To validate the results from the in vitro
assays, the effects of CEP65 on cancer cell growth and metastasis
were further investigated in vivo. Due to the low capacity
of AGS cells to form metastatic foci in animals, BICR-H1 cells were
used in the present study; these are breast cancer cells with
defined metastatic ability that have been used in a previous study
(9). CEP65 was barely detectable in
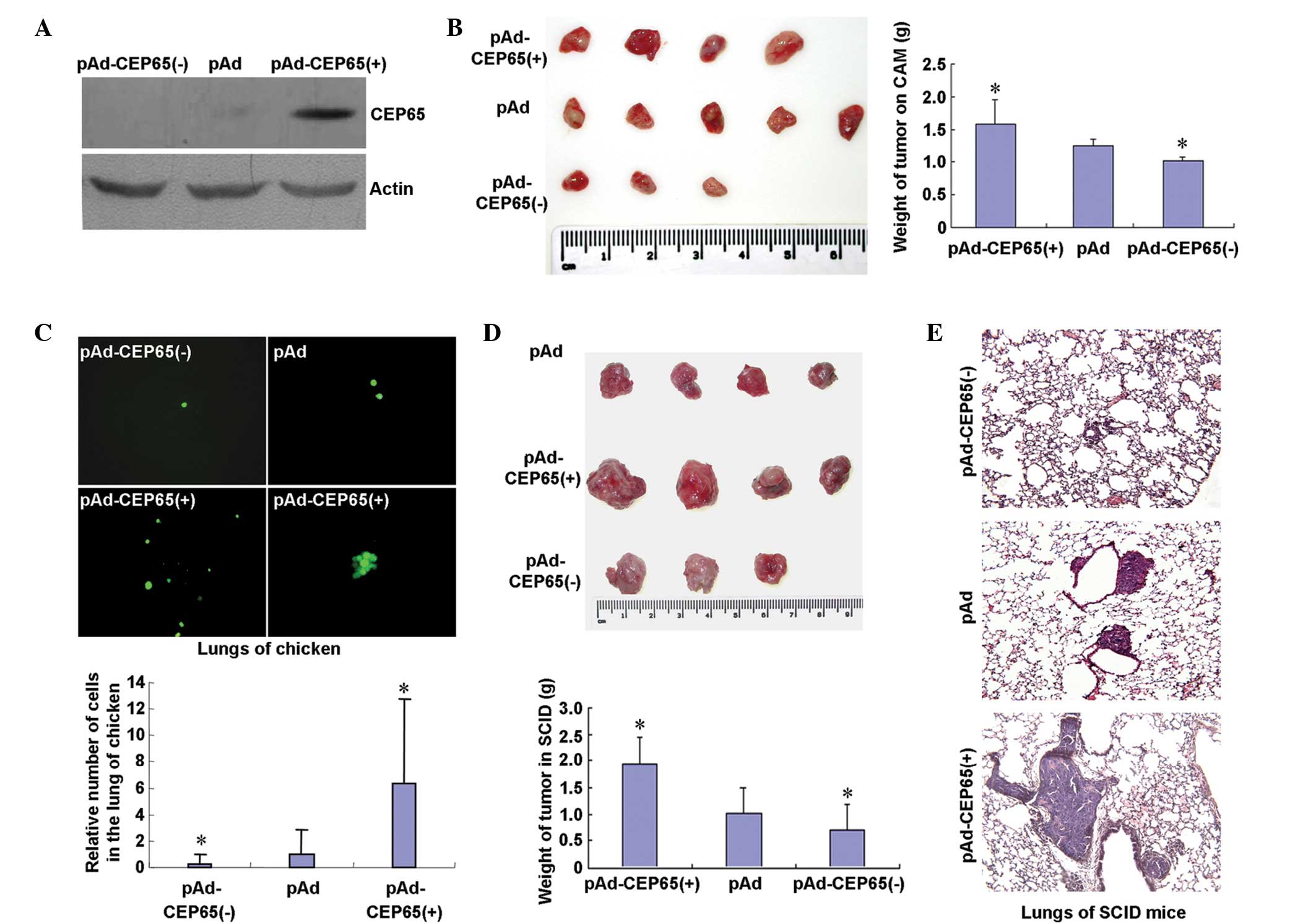
the BICR-H1 cells, using western blot analysis (Fig. 2A). Adenovirus-mediated transduction
of antisense CEP65 [pAd-CEP65(−)] resulted in the complete
elimination of CEP65 expression (Fig.
2A), while transduction of sense CEP65 [pAd-CEP65(+)] was found
to strongly increase the CEP65 protein levels (Fig. 2A). Following inoculation of the
plasmids on the chick CAMs for seven days, tumors were formed
(Fig. 2B). The tumors were weighed,
revealing that pAd-CEP65(+) increased tumor growth (P<0.05),
whereas pAd-CEP65(−) decreased tumor growth (P<0.05) (Fig. 2B). Using fluorescence microscopy,
the metastasis of the BICR-H1 cells to the chicken embryo lungs was
increased by 5.3-fold in the cells infected with pAd-CEP65(+) as
compared with the cells infected with pAd (P<0.05). Metastasis
was found to be reduced by 76.2% in the cells infected with
pAd-CEP65(−) as compared with the cells infected with pAd
(P<0.05; Fig. 2C). Furthermore,
infected BICR-H1 cells were transfected into the mammary fat pads
of SCID mice and the CEP65 expression levels were found to be
positively correlated with cell growth (Fig. 2D) and metastatic foci formation in
the lungs (Fig. 2E). These results
indicated that CEP65 promoted the growth and metastasis of the
BICR-H1 cells in vivo.
CEP65 induces MMP2 activity
The results of the present study suggested that
CEP65 has a proinvasive function in cancer cells, while the
mechanism underlying this role remains unclear. Since the
degradation of the ECM by MMPs is essential in metastasis (11), the activities of MMP2 and MMP9 in
the culture supernatant of AGS cells were investigated by
zymographic assay. As shown in Fig.
3A, the collagen-degrading activity of MMP2 was higher in the
AGS-CEP65 cell supernatant compared with the activity in the AGS
and AGS-M cells (P<0.01), however, CEP65 did not exhibit a
stimulatory effect on the activity of MMP9. The mRNA expression
levels of MMP2 and MMP9 were not affected by CEP65 (Fig. 3B). Therefore, the increased MMP2
activity in the conditioned medium of AGS-CEP65 cells did not
result from the upregulation of MMP2 expression.
Expression of CEP65 in AGS cells results
in altered gene expression
In order to understand the role of CEP65 in
regulating cancer cell invasiveness, a microarray analysis was
performed to screen genes with expression levels that were affected
by CEP65 in AGS cells. The results revealed 39 candidate genes with
>2-fold decreased expression levels and 28 genes with >2-fold
increased expression levels in the AGS-CEP65 cells.
Metastasis-associated genes, including RAP, TIMP-2 and
VTN, were among the downregulated genes. RT-PCR analysis
confirmed the decreased expression levels of these three genes in
the AGS-CEP65 cells (Fig. 3B).
Discussion
Previous studies have demonstrated that
3H11-Ag/CEP65 is highly expressed in gastric cancer tissues
(1,2). In other previous studies,
3H11-Ag/CEP65 was cloned from a cDNA library of MGC803 gastric
cancer cells (3) and CEP65 was
tagged with a red fluorescent protein (RFP), which was detected in
the nucleus and cytoplasm of COS-7 cells (6). In addition, differential extraction
has indicated that CEP65 is a potential DNA-binding protein and
peripheral membrane protein associated with nuclear envelopment.
The 150 amino acid C-terminal sequence of CEP65 may determine its
subcellular localization (6).
However, the biological role of CEP65 remains to be elucidated. In
the present study, the stimulatory effects of CEP65 in the
promotion of cancer cell growth and metastasis were identified
through in vitro and in vivo experiments, revealing
that CEP65 is a critical oncogene in tumor development.
CEP65 was found to decrease cell adhesion to
Matrigel, which was correlated with the decreased mRNA expression
level of VTN, as identified by microarray screening and RT-PCR
analysis. VTN has been identified as a cell adhesion
molecule promoting cell attachment (12–14),
therefore, the deregulation of VTN expression by CEP65 may
provide cancer cells with the capacity to detach from the ECM.
Consistent with enhanced invasiveness, CEP65 was
demonstrated to promote MMP2 activity in a zymographic assay.
However, CEP65 did not affect the mRNA expression of MMP2,
indicating that the elevated MMP2 activity may be due to altered
regulators upstream of MMP2. MMP2 belongs to a family of
Zn2+-dependent proteolytic enzymes, which are associated
with cancer cell invasion, growth, angiogenesis, inflammation and
metastasis (11,15). These enzymes are counter-balanced by
TIMPs, the endogenous antagonists of MMPs (16,17).
The TIMP family consists of at least four distinct members,
including TIMP-1, -2, -3 and -4 (16,17).
TIMP-2 is a non-glycosylated protein, which can bind to various
MMPs and antagonize their activity, exhibiting the highest
inhibitory activity against MMP2 (16). A number of studies have reported
TIMP-2-mediated inhibition of tumor growth and invasion (18–22),
while lower levels of TIMP-2 in tumor samples have been associated
with tumorigenesis (23). Blocking
TIMP-2 activity with an anti-TIMP-2 antibody has been demonstrated
to significantly induce DOV13 cell invasion due to increased
vascular endothelial growth factor (VEGF) expression (24). Another study demonstrated that, in
addition to its ability to block the action of MMPs, TIMP-2 can
also modulate tumor-host interactions, angiogenesis, tumor growth,
cell differentiation (through the regulation of cell-cycle
regulatory proteins), disruption of VEGF signaling and inhibition
of the mitogenic response of human microvascular endothelial cells
to growth factors (16), which are
MMP-independent effects. Further investigation is required to
establish whether CEP65 also functions through the aforementioned
pathways.
The present study revealed that CEP65 downregulated
RAP mRNA expression. RAP, a 39-kDa polypeptide that is
predominantly located in the endoplasmic reticulum, was identified
as a molecular chaperone that is co-purified with low density
lipoprotein receptor-related protein 1 (LRP1) (25–28).
LRP1 is involved in the catabolism of proteinases by facilitated
internalization (29). When LRP1
activity is reduced, the internalization process is impaired and
the activity of the proteinases is increased (30). These changes in activity initiate a
variety of signaling events and promote cell migration and invasion
(31). Further investigation is
required to establish whether the downregulation of RAP induced by
CEP65 is involved in enhanced cell migration and invasion.
Another critical issue that requires investigation
is how CEP65 regulates mRNA expression of TIMP-2, VTN
and RAP. CEP65 has been identified to contain eight
coiled-coil domains (3), which are
involved in the mediation of protein-protein interactions (32). To date, the spectrum of
CEP65-binding proteins is unclear, while several other centrosomal
proteins have been found to be involved in the regulation of gene
transcription (5,33). For instance, NPHP6/CEP290 is
physically associated with activating transcription factor 4
(ATF4), which may synergistically regulate renal development
(5). Considering the high homology
between CEP65 and NPHP6/CEP290, investigation of the possible
involvement of CEP65 in transcriptional regulation through ATF4 may
be useful.
In conclusion, the present study demonstrated that
CEP65 enhanced cancer cell growth, migration, invasion and
metastasis, which was correlated with the decreased expression
levels of TIMP-2, VTN and RAP. These oncogenic
roles also indicated that CEP65 may be a potential target for
cancer therapy.
Acknowledgements
The authors would like to thank Ms. Meisheng Liu and
Ms. Wei Zhao (Peking University Cancer Hospital and Institute) for
their technical assistance, and Dr. Zhiqian Zhang (Peking
University Cancer Hospital and Institute) for providing the BICR-H1
cell line. This study was supported by grants from the National 973
Program of China (nos. 2010CB529303 and 2013CB910504) and the
Natural Sciences Foundation of China (no. 30270685).
References
|
1
|
Li J, Wang Y, Wang Z and Dong Z:
Influences of amino acid sequences in FR1 region on binding
activity of the scFv and Fab of an antibody to human gastric cancer
cells. Immunol Lett. 71:157–165. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu G, Zhang M, Liu B, et al:
Radioimmunoguided surgery in gastric cancer using 131-I labeled
monoclonal antibody 3H11. Semin Surg Oncol. 10:88–94. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen DH and Shou CC: Molecular cloning of
a tumor-associated antigen recognized by monoclonal antibody 3H11.
Biochem Biophys Res Commun. 280:99–103. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jin GL, Zhang JZ, Su R, Liu XY, Wu J, Meng
L and Shou CC: Characterization of the interaction between low
density lipoprotein receptor-related protein-associated protein 1
and the cancer and embryo expression protein 65. Beijing Da Xue Xue
Bao. 37:297–301. 2005.(In Chinese). PubMed/NCBI
|
|
5
|
Sayer JA, Otto EA, O’Toole JF, Nurnberg G,
Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV,
et al: The centrosomal protein nephrocystin-6 is mutated in Joubert
syndrome and activates transcription factor ATF4. Nat Genet.
38:674–681. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guo J, Jin G, Meng L, Ma H, Nie D, Wu J,
Yuan L and Shou C: Subcellullar localization of tumor-associated
antigen 3H11Ag. Biochem Biophys Res Commun. 324:922–930. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fukasawa K: Oncogenes and tumour
suppressors take on centrosomes. Nat Rev Cancer. 7:911–924. 2007.
View Article : Google Scholar
|
|
8
|
He TC, Zhou S, da Costa LT, Yu J, Kinzler
KW and Vogelstein B: A simplified system for generating recombinant
adenoviruses. Proc Natl Acad Sci USA. 95:2509–2514. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
An P, Lei H, Zhang J, Song S, He L, Jin G,
Liu X, Wu J, Meng L, Liu M and Shou C: Suppression of tumor growth
and metastasis by a VEGFR-1 antagonizing peptide identified from a
phage display library. Int J Cancer. 111:165–173. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang H, Wang H, Shen W, Huang H, Hu L,
Ramdas L, Zhou YH, Liao WS, Fuller GN and Zhang W: Insulin-like
growth factor binding protein 2 enhances glioblastoma invasion by
activating invasion-enhancing genes. Cancer Res. 63:4315–4321.
2003.PubMed/NCBI
|
|
11
|
Hadler-Olsen E, Winberg JO and
Uhlin-Hansen L: Matrix metalloproteinases in cancer: their value as
diagnostic and prognostic markers and therapeutic targets. Tumour
Biol. 34:2041–2051. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hapke S, Kessler H, Arroyo de Prada N,
Benge A, Schmitt M, Lengyel E and Reuning U: Integrin
alpha(v)beta(3)/vitronectin interaction affects expression of the
urokinase system in human ovarian cancer cells. J Biol Chem.
276:26340–26348. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pola C, Formenti SC and Schneider RJ:
Vitronectin-αvβ3 integrin engagement directs hypoxia-resistant mTOR
activity and sustained protein synthesis linked to invasion by
breast cancer cells. Cancer Res. 73:4571–4578. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kashyap AS, Hollier BG, Manton KJ,
Satyamoorthy K, Leavesley DI and Upton Z: Insulin-like growth
factor-I:vitronectin complex-induced changes in gene expression
effect breast cell survival and migration. Endocrinology.
152:1388–1401. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chaudhary AK, Pandya S, Ghosh K and
Nadkarni A: Matrix metalloproteinase and its drug targets therapy
in solid and hematological malignancies: an overview. Mutat Res.
753:7–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stetler-Stevenson WG and Seo DW: TIMP-2:
an endogenous inhibitor of angiogenesis. Trends Mol Med. 11:97–103.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stetler-Stevenson WG and Gavil NV:
Normalization of the tumor microenvironment: evidence for tissue
inhibitor of metalloproteinase-2 as a cancer therapeutic. Connect
Tissue Res. 55:13–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brand K, Baker AH, Perez-Cantó A, Possling
A, Sacharjat M, Geheeb M and Arnold W: Treatment of colorectal
liver metastases by adenoviral transfer of tissue inhibitor of
metalloproteinases-2 into the liver tissue. Cancer Res.
60:5723–5730. 2000.PubMed/NCBI
|
|
19
|
Zhao YG, Xiao AZ, Park HI, Newcomer RG,
Yan M, Man YG, Heffelfinger SC and Sang QX: Endometase/matrilysin-2
in human breast ductal carcinoma in situ and its inhibition by
tissue inhibitors of metalloproteinases-2 and -4: a putative role
in the initiation of breast cancer invasion. Cancer Res.
64:590–598. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu W, Zhou X, Hong B, Liu J and Yue Z:
Suppression of invasion in human U87 glioma cells by
adenovirus-mediated co-transfer of TIMP-2 and PTEN gene. Cancer
Lett. 214:205–213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee YK, So IS, Lee SC, Lee JH, Lee CW, Kim
WM, Park MK, Lee ST, Park DY, Shin DY, et al: Suppression of
distant pulmonary metastasis of MDA-MB 435 human breast carcinoma
established in mammary fat pads of nude mice by retroviral-mediated
TIMP-2 gene transfer. J Gene Med. 7:145–157. 2005. View Article : Google Scholar
|
|
22
|
Lin CW, Chen PN, Chen MK, Yang WE, Tang
CH, Yang SF and Hsieh YS: Kaempferol reduces matrix
metalloproteinase-2 expression by down-regulating ERK1/2 and the
activator protein-1 signaling pathways in oral cancer cells. PLoS
One. 8:e808832013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Galm O, Suzuki H, Akiyama Y, Esteller M,
Brock MV, Osieka R, Baylin SB and Herman JG: Inactivation of the
tissue inhibitor of metalloproteinases-2 gene by promoter
hypermethylation in lymphoid malignancies. Oncogene. 24:4799–4805.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang FQ, So J, Reierstad S and Fishman DA:
Vascular endothelial growth factor-regulated ovarian cancer
invasion and migration involves expression and activation of matrix
metalloproteinases. Int J Cancer. 118:879–888. 2006. View Article : Google Scholar
|
|
25
|
Herz J, Hamann U, Rogne S, Myklebost O,
Gausepohl H and Stanley KK: Surface location and high affinity for
calcium of a 500-kd liver membrane protein closely related to the
LDL-receptor suggest a physiological role as lipoprotein receptor.
EMBO J. 7:4119–4127. 1988.PubMed/NCBI
|
|
26
|
Strickland DK, Ashcom JD, Williams S,
Burgess WH, Migliorini M and Argraves WS: Sequence identity between
the alpha 2-macroglobulin receptor and low density lipoprotein
receptor-related protein suggests that this molecule is a
multifunctional receptor. J Biol Chem. 265:17401–17404.
1990.PubMed/NCBI
|
|
27
|
Willnow TE, Rohlmann A, Horton J, Otani H,
Braun JR, Hammer RE and Herz J: RAP, a specialized chaperone,
prevents ligand-induced ER retention and degradation of LDL
receptor-related endocytic receptors. EMBO J. 15:2632–2639.
1996.PubMed/NCBI
|
|
28
|
Bu G: Receptor-associated protein: a
specialized chaperone and antagonist for members of the LDL
receptor gene family. Curr Opin Lipidol. 9:149–155. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Herz J and Strickland DK: LRP: a
multifunctional scavenger and signaling receptor. J Clin Invest.
108:779–784. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamamoto K, Troeberg L, Scilabra SD,
Pelosi M, Murphy CL, Strickland DK and Nagase H: LRP-1-mediated
endocytosis regulates extracellular activity of ADAMTS-5 in
articular cartilage. FASEB J. 27:511–521. 2013. View Article : Google Scholar :
|
|
31
|
Mantuano E, Lam MS and Gonias SL: LRP1
assembles unique co-receptor systems to initiate cell signaling in
response to tissue-type plasminogen activator and myelin-associated
glycoprotein. J Biol Chem. 288:34009–34018. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Woolfson DN, Bartlett GJ, Bruning M and
Thomson AR: New currency for old rope: from coiled-coil assemblies
to α-helical barrels. Curr Opin Struct Biol. 22:432–441. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koyanagi M, Hijikata M, Watashi K, Masui O
and Shimotohno K: Centrosomal P4.1-associated protein is a new
member of transcriptional coactivators for nuclear factor-kappaB. J
Biol Chem. 280:12430–12437. 2005. View Article : Google Scholar : PubMed/NCBI
|