Introduction
Glioblastoma (GBM) is the most common type of brain
tumor, with high mortality rates. The aggressive biology of GBM and
resistance to therapy are major clinical problems for patients
diagnosed with the disease (1). At
present, standard treatment includes maximal safe surgery,
concomitant radiotherapy and temozolomide (TMZ) chemotherapy, where
a median survival time of <15 months is observed in patients fit
for this current first-line therapy (1). In terms of years of life lost, the
population burden from GBM is the highest of all malignancies
(2). GBM frequently affects young
people (30–40 years of age), as well as the elderly (3). Physically or cognitively disabled
patients require care from the early stages of the disease, which
results in increased expenses. Despite advances in therapeutic
strategies, no significant reductions in the age-standardised
incidence and mortality rates of this disease have been observed
within the last decade (3).
Patients with a good Eastern Cooperative Oncology
Group performance status or Karnofsky score at the time of
diagnosis undergo surgical resection (4,5). Complete
removal of the tumor is challenging due to the infiltrative nature
of GBM; however, 80–95% debulking is typically achieved unless the
disease is in a neurologically eloquent location (6). The standard treatment, known as ‘The
Stupp Protocol’, consists of concurrent temozolomide (TMZ) therapy
and radiotherapy (60 Gy/30 fractions), followed by subsequent
adjuvant TMZ therapy for 6–12 cycles (1). TMZ functions by alkylating the guanine
residues in DNA, resulting in mispairing with thymine during DNA
replication and cellular arrest in rapidly dividing cells (7,8).
Methylguanine-DNA methyltransferase (MGMT) removes
mutagenic alkyl adducts and, thus, protects DNA from the damage
induced by TMZ and other alkylating agents. Loss of MGMT expression
commonly occurs in human neoplasia (9). The expression of MGMT is regulated by
hypermethylation of the CpG islands within the promoter and
enhancer regions of the gene (10–12). MGMT
deficiency is presumed in GBMs with a methylated MGMT
promoter, resulting in the enhanced cytotoxicity of TMZ (13). Studies have demonstrated that presence
of MGMT promoter methylation is correlated with the response
of patients to treatment with TMZ; thus, it is considered to be a
predictive biomarker in TMZ-treated GBMs, even in elderly patients
(14,15). Between 25 and 50% of GBMs are
methylated at the MGMT promoter; therefore, using
MGMT methylation status clinically as a predictive biomarker
for GBM patients has received increased attention, with the
long-term aim to use it as a biomarker to assign alkylating therapy
to individual patients. However, a number of studies have reported
discordance between MGMT methylation status and treatment
outcome, and no clear alternative treatment is available for
patients with unmethylated MGMT tumors, who derive only
limited benefit from TMZ (13).
Based on the signatory gene expression profiles
using data generated from The Cancer Genome Atlas (TCGA) (16), four distinct molecular subtypes of GBM
were described: Classical, mesenchymal, neural and proneural
(17). These subtypes have also been
associated with characteristic genetic alterations (17). The classical subtype is associated
with an astrocytic expression profile, with frequent epidermal
growth factor receptor (EGFR) amplification, concomitant
chromosome 7 amplification and chromosome 10 loss, and focal
deletions of 9p encompassing cyclin-dependent kinase inhibitor 2A
(CDKN2A). Notably, the classical subtype is not associated
with TP53 mutations, which are common in GBMs (17). The mesenchymal subtype is typified by
the expression of mesenchymal markers, with frequent deletions or
mutations of the neurofibromin 1 (NF1) and phosphatase and
tensin homolog (PTEN) genes. The neural subtype exhibits
expression of neuronal markers and displays various mutations and
copy number alterations, including amplification of EGFR and
deletion of PTEN. The proneural subtype exhibits an
oligodendrocytic expression signature and is characterized by
TP53 mutations. In addition, focal amplifications of the
chromosome 4q12 region, which contains the oligodendrocytic
development gene α-type platelet-derived growth factor receptor
(PDGFRA), or mutations of the isocitrate dehydrogenase 1
gene (IDH1) are observed. The TCGA group has recently
published an updated comprehensive genomic, epigenomic,
transcriptomic and proteomic analysis of >500 GBMs (18). Patients were categorized into the four
different groups and the overall survival was stratified based on
MGMT methylation. Notably, the findings indicated that
MGMT methylation may be a predictive biomarker for response
to treatment, but only in the classical subtype of GBM.
In the present study, MGMT methylation and
somatic mutations were assessed using semiconductor sequencing
technology in the same tumor DNA. The cohort included biopsies from
35 GBM patients, who had been treated uniformly with standard
concurrent radiotherapy plus TMZ. The association between
MGMT methylation status/mutation profile and survival was
also evaluated.
Materials and methods
Patient samples
Formalin-fixed paraffin-embedded tissues from
patients with GBM, which had been obtained during the initial
surgical resection, were collected from the Australian Genomics and
Clinical Outcomes of Glioma biobank (www.agogbio.org.au). All the patients provided written
informed consent and the study was approved by the Institutional
Human Research Ethics Committee of South Eastern Sydney Local
Health District (SESLD) Human Research Ethics Committee (HREC),
University of New South Wales (Sydney, Australia). Specimens were
obtained prior to any chemotherapy or radiotherapy, and the patient
information, including age, gender, extent of tumor removal,
pathology and treatment, was recorded. Only patients receiving
maximal debulking of the tumor (>85%) and undergoing concurrent
chemoradiotherapy (60 Gy in 30 fractions with 75
mg/m2/day TMZ) were included in this study. The number
of adjuvant cycles of TMZ, determined by clinical variables
including toxicity, response to treatment and progression, was
noted. Time to progression (TTP; measured from the start of
chemoradiotherapy to the time of clinical progression) was
determined by the treating oncologist (Professor Anna Nowak).
MGMT promoter methylation
Bisulfite modification prior to CpG pyrosequencing
was performed to assess the percentage level of MGMT
promoter methylation for each tumor specimen, as described
previously (19). Chemically
methylated or unmethylated universal human genomic DNA (EMD
Millipore, Billerica, MA, USA) controls were included with each
batch. The CpG pyrosequencing methylation assay was performed using
the PyroMarkTM MGMT kit (Qiagen, Alameda, CA, USA) on a PSQTM96 MA
system (Qiagen) according to the manufacturer's instructions.
Detection of EGFR gene
amplification
EGFR amplification was recorded in the
patient pathology reports by PathWest Laboratory Medicine
(Nedlands, Australia) using the Multiplex Ligation-dependent Probe
Amplification assay (MLPA; MCR-Holland, Amsterdam, Netherlands), as
described previously (20).
Targeted resequencing of DNA
In order to amplify the target regions of 50
oncogenes and tumor suppressor genes that are implicated in solid
tumors, amplicon libraries for individual patient DNA samples were
prepared using the Ion AmpliSeq Cancer Hot Spot Panel v2 (Life
Technologies, Grand Island, NY, USA), as described previously
(21). Subsequently, semi-conductor
sequencing was performed using the Ion Personal Genome Machine
(PGM) 200 Sequencing kit (Life Technologies) and the Ion PGM
Sequencer (21). Torrent Suite™
v3.6.2 software (Life Technologies) was used to parse barcoded
reads, align reads to the reference genome (human genome build 19),
base call and generate run metrics, including the chip loading
efficiency, total read counts, quality and total coverage. In
addition, the ANNOVAR™ (Biobase Biological Databases; http://www.biobase-international.com)
and Oncomine Gene Browser (Life Technologies) software were used to
identify variants, as well as predict amino acid changes and
clinical significance. The Integrative Genomics Viewer (Broad
Institute, Cambridge, MA, USA; http://www.broadinstitute.org/igv/) was used to
visualize the alignment of the reads, detect the presence of
variants against the reference genome and verify the integrity of
variant calls by detecting possible strand bias and sequencing
errors (21).
Statistical analysis
The primary end point was the overall survival. A
Kaplan-Meier survival analysis was used to generate survival curves
and estimates of median survival periods. All statistical analyses
were performed using SPSS software (version 22.0; SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Patient characteristic
A total of 35 patients with a diagnosis of GBM were
included in the current study (Table
I). The median age was 58 years (range, 31–80 years) and the
study included 25 males and 10 females. The median overall survival
of the cohort was 13.0 months [95% confidence interval (CI),
9.7–16.3]. The median TTP of the cohort was 6.0 months (95% CI,
4.7–7.3).
 | Table I.Summary of patient characteristics
and treatment results. |
Table I.
Summary of patient characteristics
and treatment results.
| Characteristic | Value |
|---|
| Patients, n | 35 |
| Age, years |
|
|
Median | 58 |
|
Range | 30.5–79.6 |
| Gender, n |
|
|
Male | 25 |
|
Female | 10 |
| Overall survival,
months |
|
|
Median | 13 |
|
95%CI | 9.7–16.3 |
| Time to
progression, months |
|
|
Median | 6 |
| 95%
CI | 4.7–7.3 |
The MGMT promoter methylation status was
detected in 33 out of the 35 patients, with 16 tumors (48%) found
to be methylated at the MGMT promoter region. Tumor
MGMT methylation was associated with significantly longer
survival rates (median survival, 29.0 months; 95% CI, 20.1–32.9)
compared with patients without MGMT methylated tumors
(median survival, 12.0 months; 95% CI, 10.1–13.9; log-rank P=0.025;
Fig. 1A). A strong association was
also observed between MGMT methylation and longer TTP, since
the median TTP for patients with methylated MGMT tumors was
13.2 months (95% CI, 4.1–19.9) compared with 5.6 months (95% CI,
4.5–7.5) for patients with an unmethylated tumor MGMT status
(log-rank P=0.011; Fig. 1B).
Overall gene alterations
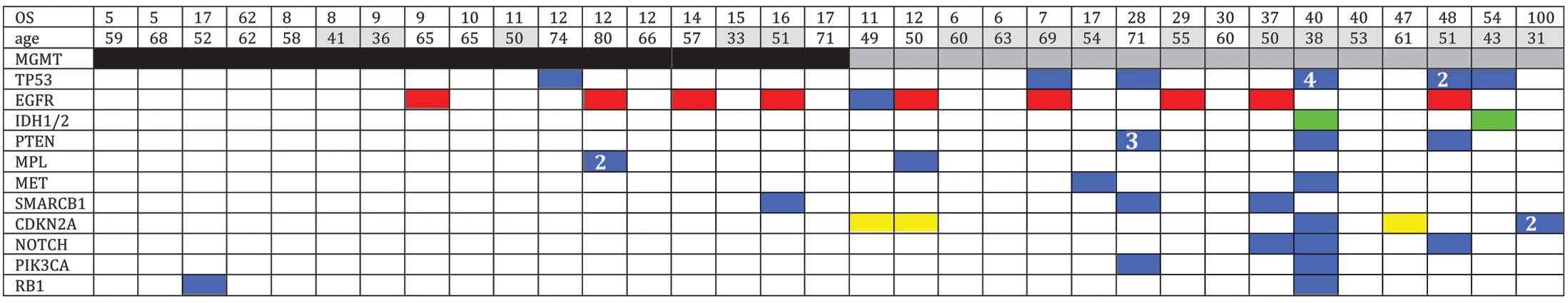
Of the 35 specimens sequenced, multiple (>2)
non-synonymous mutations were detected in 10 specimens. In the
present study, mutations were identified in the TP53 (20%),
CDKN2A (11%) and PTEN (9%) genes, as well as
mutations and/or amplifications in the EGFR gene (29%). In
addition, two tumors exhibited mutations in IDH1. Mutations
in MPL (6%), SMARCB1 (9%), NOTCH1 (9%),
PIK3CA (9%), MET (6%) and RB1 (6%) were also
detected (Fig. 2).
The mutation profiles were mapped according to
MGMT promoter methylation (Fig.
2), which revealed that non-synonymous mutations were more
frequent in the MGMT methylated GBMs compared with the
MGMT unmethylated GBMs. Furthermore, mutations in the
CDKN2A, PTEN and PIKC3A genes were only
identified in MGMT methylated GBMs. Amplification and/or
mutation in the EGFR gene and deletion of CDKN2A are
hallmarks of the classical tumor subtype. A recent study has
reported that the predictive value of MGMT methylation was
only applicable in the classical subtype (15). In the current study, the frequency of
EGFR/CDKN2A gene aberrations was found to be higher
in the MGMT methylated group of tumors (56%) when compared
with the MGMT unmethylated tumors (24%).
Discussion
The presence of MGMT promoter methylation in
GBM is a predictor of response to TMZ treatment and of overall
survival. In the present study, the mutations detected in
MGMT methylated GBMs were four times higher compared with
MGMT unmethylated GBMs. This observation may be attributed
to a lack of DNA repair function upon presence of lower levels of
the MGMT protein.
In 2005, MGMT promoter methylation was
reported to be associated with a survival benefit from alkylating
agent chemotherapy in GBM patients, in a companion study based on a
phase III concurrent radiation therapy and TMZ trial (13). A number of studies have demonstrated
an even higher survival benefit in patients treated with alkylating
agents, who presented tumors with MGMT promoter methylation,
compared with patients having tumors with an unmethylated
MGMT promoter (13,22,23).
Therefore, MGMT promoter methylation is a favorable
prognostic marker, in addition to a favorable predictive marker for
response to TMZ treatment in patients with GBM. The molecular basis
for the differential response to TMZ treatment in patients is well
established. Alkylating drugs, including TMZ, induce DNA damage by
introducing alkyl adducts into the DNA molecules. This causes
genetic mutations and cross-links between DNA strands that inhibit
DNA replication and thereby trigger cell death. However,
MGMT removes the alkyl adducts from the DNA as they are
introduced, preventing subsequent mutational damage. Tumor cells
expressing MGMT are thus resistant to alkylating drugs (24).
In the current study, the TTP following
chemoradiotherapy and subsequent overall survival were
significantly increased in patients with MGMT methylation
compared with patients without methylated tumors. Thus, the present
study confirmed the role of MGMT methylation and its
associated sensitivity to TMZ (25,26).
The current study investigated the prevalence and
types of genomic alterations in GBM and whether these correlate
with MGMT methylation. The results demonstrated that 75% of
MGMT methylated tumors harbored one or more somatic
mutations, compared with 35% of unmethylated cases. Overall, the
mutations identified in MGMT methylated were four times
higher compared with MGMT unmethylated tumors. Notably,
mutations were identified in the TP53, CDKN2A,
PTEN and PIK3CA genes in MGMT methylated
cases. Since these cases also exhibited improved overall survival,
further investigations should determine whether these genomic
aberrations work synergistically with MGMT methylation
status and contribute to the response to treatment and overall
prognosis. MGMT methylation may play a dual role, increasing
sensitivity to TMZ and increasing genetic instability, particularly
by facilitating the appearance of G to A transition mutations. The
TCGA dataset of GBMs revealed increased frequencies of TP53
and PTEN point mutations in MGMT methylated GBMs
compared with MGMT unmethylated GBMs (16). In the current study, mutations in
PTEN and TP53 were also found to be associated with
MGMT promoter methylation, confirming the findings of the
TCGA. MGMT methylation is commonly associated with G:C to
A:T mutations in KRAS and TP53 genes in colorectal
cancer (27,28). Therefore, MGMT promoter
methylation may be an early event in the development of methylated
GBM, preceding other characteristic mutations in this subgroup.
In the present study, higher frequencies of
EGFR and CDKN2A alterations were observed in the
MGMT methylation group. Although the tumors were not
categorized into proneural, neural, mesenchymal or classical
subtypes (17), the higher
frequencies of EGFR/CDKN2A indicate that the
MGMT methylation group may be enriched for the classical
subtype. Furhtermore, the current study is in accordance with the
findings of Brennan et al (18), demonstrating that the classical
subtype is more responsive to TMZ, which is reflected by the longer
TTP and overall survival observed.
In conclusion, MGMT methylation was strongly
associated with the response to TMZ treatment and overall survival
in GBMs, as previously reported. Methylation was also found to be
associated with a significantly higher mutation rate. Therefore, it
is hypothesized that methylation may be one of the earliest
preneoplastic events in this subgroup. A larger cohort of patients
is required to further investigate the association of MGMT
methylation and specific mutations (including those in EGFR,
CDKN2A and TP53) with overall survival in TMZ-treated
GBMs.
References
|
1
|
Stupp R, Mason WP, van den Bent MJ, et al:
European Organisation for Research and Treatment of Cancer Brain
Tumor and Radiotherapy Groups; National Cancer Institute of Canada
Clinical Trials Group: Radiotherapy plus concomitant and adjuvant
temozolomide for glioblastoma. N Engl J Med. 352:987–996. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burnet NG, Jefferies SJ, Benson RJ, et al:
Years of life lost (YLL) from cancer is an important measure of
population burden – and should be considered when allocating
research funds. Br J Cancer. 92:241–245. 2005.PubMed/NCBI
|
|
3
|
Leibetseder A, Ackerl M, Flechl B, et al:
Outcome and molecular characteristics of adolescent and young adult
patients with newly diagnosed primary glioblastoma: a study of the
Society of Austrian Neurooncology (SANO). Neuro Oncol. 15:112–121.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kinsella TJ, Collins J, Rowland J, et al:
Pharmacology and phase I/II study of continuous intravenous
infusions of iododeoxyuridine and hyperfractionated radiotherapy in
patients with glioblastoma multiforme. J Clin Oncol. 6:871–879.
1988.PubMed/NCBI
|
|
5
|
Hammoud MA, Sawaya R, Shi W, Thall PF and
Leeds NE: Prognostic significance of preoperative MRI scans in
glioblastoma multiforme. J Neurooncol. 27:65–73. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gulati S, Jakola AS, Nerland US, Weber C
and Solheim O: The risk of getting worse: surgically acquired
deficits, perioperative complications, and functional outcomes
after primary resection of glioblastoma. World Neurosurg.
76:572–579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Friedman HS, Kerby T and Calvert H:
Temozolomide and treatment of malignant glioma. Clin Cancer Res.
6:2585–2597. 2000.PubMed/NCBI
|
|
8
|
Roos WP and Kaina B: DNA damage-induced
apoptosis: from specific DNA lesions to the DNA damage response and
apoptosis. Cancer Lett. 332:237–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Esteller M, Hamilton SR, Burger PC, et al:
Inactivation of the DNA repair gene O6-methylguanine-DNA
methyltransferase by promoter hypermethylation is a common event in
primary human neoplasia. Cancer Res. 59:793–797. 1999.PubMed/NCBI
|
|
10
|
Felsberg J, Thon N, Eigenbrod S, et al:
German Glioma Network: Promoter methylation and expression of MGMT
and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in
paired primary and recurrent glioblastomas. Int J Cancer.
129:659–670. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Esteller M: Epigenetic lesions causing
genetic lesions in human cancer: promoter hypermethylation of DNA
repair genes. Eur J Cancer. 36:2294–2300. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Esteller M: CpG island hypermethylation
and tumor suppressor genes: a booming present, a brighter future.
Oncogene. 21:5427–5440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hegi ME, Diserens AC, Gorlia T, et al:
MGMT gene silencing and benefit from temozolomide in glioblastoma.
N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reifenberger G, Hentschel B, Felsberg J,
et al: German Glioma Network: Predictive impact of MGMT promoter
methylation in glioblastoma of the elderly. Int J Cancer.
131:1342–1350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stummer W, Nestler U, Stockhammer F, et
al: Favorable outcome in the elderly cohort treated by concomitant
temozolomide radiochemotherapy in a multicentric phase II safety
study of 5-ALA. J Neurooncol. 103:361–370. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Verhaak RG, Hoadley KA, Purdom E, et al:
Cancer Genome Atlas Research Network: Integrated genomic analysis
identifies clinically relevant subtypes of glioblastoma
characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1.
Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brennan CW, Verhaak RG, McKenna A, et al:
TCGA Research Network: The somatic genomic landscape of
glioblastoma. Cell. 155:462–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McDonald KL, Rapkins RW, Olivier J, et al:
The T genotype of the MGMT C>T (rs16906252) enhancer
single-nucleotide polymorphism (SNP) is associated with promoter
methylation and longer survival in glioblastoma patients. Eur J
Cancer. 49:360–368. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jeuken J, Sijben A, Alenda C, et al:
Robust detection of EGFR copy number changes and EGFR variant III:
technical aspects and relevance for glioma diagnostics. Brain
Pathol. 19:661–671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tabone T, Abuhusain HJ, Nowak AK, et al:
Multigene profiling to identify alternative treatment options for
glioblastoma: a pilot study. J Clin Pathol. 67:550–555. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Esteller M, Garcia-Foncillas J, Andion E,
et al: Inactivation of the DNA-repair gene MGMT and the clinical
response of gliomas to alkylating agents. N Engl J Med.
343:1350–1354. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hegi ME, Diserens AC, Godard S, et al:
Clinical trial substantiates the predictive value of
O-6-methylguanine-DNA methyltransferase promoter methylation in
glioblastoma patients treated with temozolomide. Clin Cancer Res.
10:1871–1874. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pegg AE: Mammalian O6-alkylguanine-DNA
alkyltransferase: regulation and importance in response to
alkylating carcinogenic and therapeutic agents. Cancer Res.
50:6119–6129. 1990.PubMed/NCBI
|
|
25
|
Christians A, Hartmann C, Benner A, et al:
Prognostic value of three different methods of MGMT promoter
methylation analysis in a prospective trial on newly diagnosed
glioblastoma. PloS one. 7:e334492012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karayan-Tapon L, Quillien V, Guilhot J, et
al: Prognostic value of O6-methylguanine-DNA methyltransferase
status in glioblastoma patients, assessed by five different
methods. J Neurooncol. 97:311–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rosty C, Young JP, Walsh MD, et al: PIK3CA
activating mutation in colorectal carcinoma: associations with
molecular features and survival. PLoS One. 8:e654792013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shen L, Kondo Y, Rosner GL, et al: MGMT
promoter methylation and field defect in sporadic colorectal
cancer. J Natl Cancer Inst. 97:1330–1338. 2005. View Article : Google Scholar : PubMed/NCBI
|