Introduction
Lung cancer is a leading cause of cancer-associated
mortality worldwide and the most common type is non-small cell lung
cancer (NSCLC) (1). Despite progress
in the development of molecular-targeted therapeutic agents and
surgical approaches, chemotherapy remains an important strategy for
the treatment of NSCLC. A combination of a next-generation
cytotoxic agents and a platinum compound is the standard treatment
regimen for patients with advanced NSCLC (2,3). The
cytotoxicity of cisplatin (CDDP), a platinum compound used in
chemotherapy, is predominantly due to its interaction with DNA,
forming DNA adducts that result in the activation of a number of
apoptosis signaling pathways (4).
However, the resistance of NSCLC cells to CDDP, which is currently
a barrier in the treatment of patients with NSCLC, is associated
with inactivation of these apoptosis signaling pathways (5,6).
In tumor cells, numerous apoptosis-associated genes
appear to have methylated CpG islands. DNA methylation is a crucial
regulator in various biological processes; however, aberrant CpG
island hypermethylation in gene promoter regions may impact the
cell cycle, proliferation, apoptosis, metastasis, drug resistance
and intracellular signaling. Therefore, the occurrence of this
aberrant hypermethylation is important in carcinogenesis and cancer
treatment. DNA hypermethylation is an epigenetic modification that
leads to the transcriptional silencing of regulatory genes in
various types of human cancer. Furthermore, DNA methyltransferase
(DNMT) enzymes are critical for the maintenance of normal DNA
methylation (7,8).
The expression of apoptosis-associated genes, which
is silenced by aberrant DNA hypermethylation, can be restored by
the pyrimidine nucleoside analog of cytosine, 5-aza-2′deoxycytidine
(DAC) (9,10). The loss of DNA hypermethylation occurs
through the formation of a covalent complex between DAC and DNMTs
(11). Therefore, the clinical
application of DAC in the treatment of patients with
myelodysplastic syndrome (MDS) resulted in significant
demethylation of apoptosis-associated genes in certain cases
(12).
The aim of the present study was to evaluate the
effect of combined therapy with DAC and CDDP on the human lung
adenocarcinoma cell line, P15. In addition, the mRNA expression
levels of six associated genes, including p73, B-cell
lymphoma (Bcl)-xL; Bcl-2-associated agonist of cell
death (BAD); Bcl-2-associated X protein (Bax),
p14ARF and p16INK4a, were
analyzed.
Materials and methods
Cell culture and DAC treatment
The P15 cell line, which is derived from lung
adenocarcinoma, was obtained from the Center for Experimental
Animals of Sun Yat-sen University (Guangzhou, China). P15 cells
were maintained in RPMI-1640 medium (Corning, Inc., Corning, New
York, NY, USA) supplemented with 10% fetal bovine serum in an
atmosphere of 5% CO2 at 37°C. The P15 cell culture was
exposed to different concentrations of the demethylation agent, DAC
(2, 5 and 10 µmol/l; Sigma-Aldrich, St. Louis, MO, USA), for 72 h
to assess growth inhibition and monitor the mRNA expression levels
of specific genes.
Clone formation assay
The exponentially growing cells were trypsinized
(Sigma-Aldrich) into a single cell suspension following treatment
with 5 µM DAC for 72 h. Next, the cells were inoculated into 6-well
plates, with each well containing 500 cells. The cells were treated
with 0.05 µM CDDP for 72 h and cultured at 37°C in an atmosphere of
5% CO2 to facilitate colony formation for 12 days. After
rinsing twice with phosphate-buffered saline (PBS), the colonies
were fixed with 1 ml methanol for 30 min. The fixing solution was
discarded and 1 ml Giemsa dye (Wuhan Huamei Biotechnology Co.,
Ltd., Beijing, China) was applied for 30 min to stain the cells.
Colonies containing a minimum of 50 viable cells were counted using
an inverted fluorescence microscope (Olympus BX-51; Olympus
Corporation, Tokyo, Japan) to quantify the survival fractions. The
survival fraction was calculated using the following formula:
Survival fraction = number of colonies/500 cells.
Growth inhibitory activities of
DAC
Cells (0.25×104) were seeded in
triplicate in 24-well plates two days prior to treatment (day −2).
The seeded cells were divided into four groups as follows:
untreated control; DAC alone; CDDP alone; and DAC followed by CDDP
treatment groups. New medium containing 5 µM of DAC (or PBS) was
added to the appropriate groups on day 0. On day 3, the cells were
rinsed with PBS and new medium containing 0.05 µM CDDP was added to
the CDDP alone and DAC followed by CDDP treatment groups, according
to the manufacturer's instructions. The cells were digested with
0.25% trypsin and 0.5 M EDTA and counted daily from seeding until
day 6.
Total RNA extraction and reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from the cells using TRIzol
reagent (Omega Bio-Tek, Inc., Norcross, GA, USA), purified and
dissolved in diethylpyrocarbonate-treated distilled water. Next,
1.0 µg total RNA was reverse-transcribed using avian myeloblastosis
virus reverse transcriptase (Takara Biotechnology Co., Ltd.,
Dalian, China). cDNA was then obtained and PCR amplification was
performed using the primers indicated in Table I, under the following conditions:
Initial denaturation at 94°C for 2 min, followed by 30–35 cycles of
denaturation at 94°C for 30 sec, annealing at 55–63°C for 30 sec
and extension at 72°C for 1 min. The PCR mixture consisted of:
distilled H2O 12.5 µl, 5X PCR buffer 2 µl (Takara
Biotechnology Co., Ltd.), Taq enzyme 0.5 µl (Takara Biotechnology
Co., Ltd.), NTPs 2 µl (Takara Biotechnology Co., Ltd.), forward
primer 1 µl, reverse primer 1 µl, cDNA 1 µl. The primers were
designed and synthesized by Takara Biotechnology Co. Ltd. The PCR
products were electrophoresed on 3% agarose gels, stained with
ethidium bromide (Takara Biotechnology Co., Ltd.) and visualized
under ultraviolet illumination using a Uvidoc gel documentation
system (GelDoc XR, Bio-Rad Laboratories, Inc., Hercules, CA, USA).
β-actin was selected as the endogenous reference gene. The
six apoptosis-related genes, such as p73,
Bcl-xL, BAD, Bax,
p14ARF, and p16INK4a, were
investigated by RT-PCR.
 | Table I.Genes and primer sequences. |
Table I.
Genes and primer sequences.
|
| Nucleotide
sequence, 5′-3′ |
|
|
|---|
|
|
|
|
|
|---|
| Gene | Forward primer | Reverse primer | Annealing
temperature, °C | Product size,
bp |
|---|
| β-actin |
CCTTCCTGGGCATGGAGTCCT |
AATCTCATCTTGTTTTCTGCG | 55 | 407 |
| p73 |
ACTTCAACGAAGGACAGTCTGCT |
AATTCCGTCCCCACCTGTG | 63 | 142 |
|
Bcl-xL |
CATGGCAGCAGTAAAGCAAG |
GCATTGTTCCCATAGAGTTCC | 55 | 351 |
| BAD |
TTTAAGAAGGGACTTCCTCGCC |
GAGCTTCCCCTGCCCAAGTT | 60 | 119 |
| Bax |
ATCCAGGATCGAGCAGGGCG |
ACTCGCTCAGCTTCTTGGTG | 58 | 94 |
|
p14ARF |
GAGTGAGGGTTTTCGTGG |
GCCCATCATCATGACCTG | 55 | 153 |
|
p16INK4a |
CAACGCACCGAATAGTTACGG |
GCGCAGTTGGGCTCCG | 55 | 105 |
Terminal deoxynucleotidyl transferase
(TdT)-mediated dUTP nick end labeling (TUNEL) analysis
TUNEL (Roche Diagnostics, Basel, Switzerland) was
used to evaluate the treatment-associated apoptosis, according to
the manufacturer's instructions. Cells (0.25×104/well)
were seeded on the slides in 6-well plates and exposed to 5 µM DAC
for 72 h, followed by 1.25 µM CDDP for 72 h. The slides were washed
with PBS, fixed for 60 min with 4% paraformaldehyde and incubated
with TUNEL detection solution containing TdT and fluorescein
isothiocyanate (FITC)-labeled solution. DNA strand breaks were
labeled with FITC-conjugated dUTP and the nuclei were visualized
using an inverted fluorescence microscope (Olympus BX-51; Olympus
Corporation).
Statistical analysis
Statistical analyses were performed using the SPSS
software (version 16.0; SPSS Inc., Chicago, IL, USA). Survival and
apoptosis were analyzed using Welch's t-test and P<0.05 was
considered to indicate a statistically significant difference. Data
are presented as the mean ± standard deviation.
Results
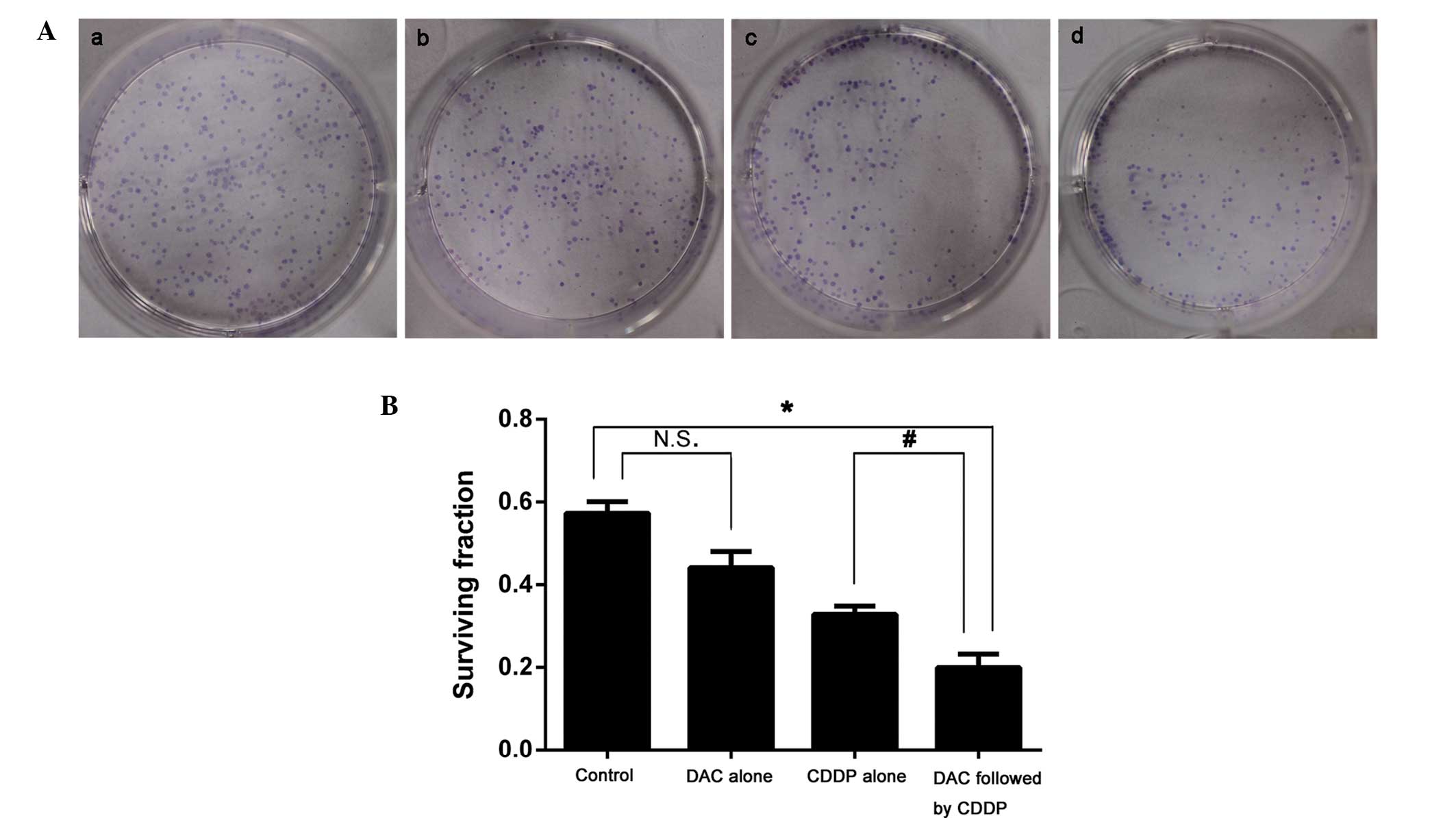
Colony formation
The survival fraction for colony formation in cells
treated with DAC alone (0.440±0.009) was not significantly
different from that of the control group (0.574±0.004; P>0.05).
However, following combined treatment with DAC and CDDP, the
survival fraction for colony formation was significantly decreased
when compared with the DAC alone group (0.200±0.007 vs.
0.328±0.007; P<0.05; Fig. 1).
Cell proliferation inhibition
The growth curve of cell proliferation following
treatment with DAC and/or CDDP is indicated in Fig. 2. On day 6, the number of cells in the
combined treatment group was 15% lower compared with the number of
cells in the control group (P=0.004). Furthermore, cell
proliferation was significantly more suppressed in DAC alone group
compared with the control group, as well as in the combined DAC and
CDDP treatment group compared with the CDDP alone group
(P<0.05).
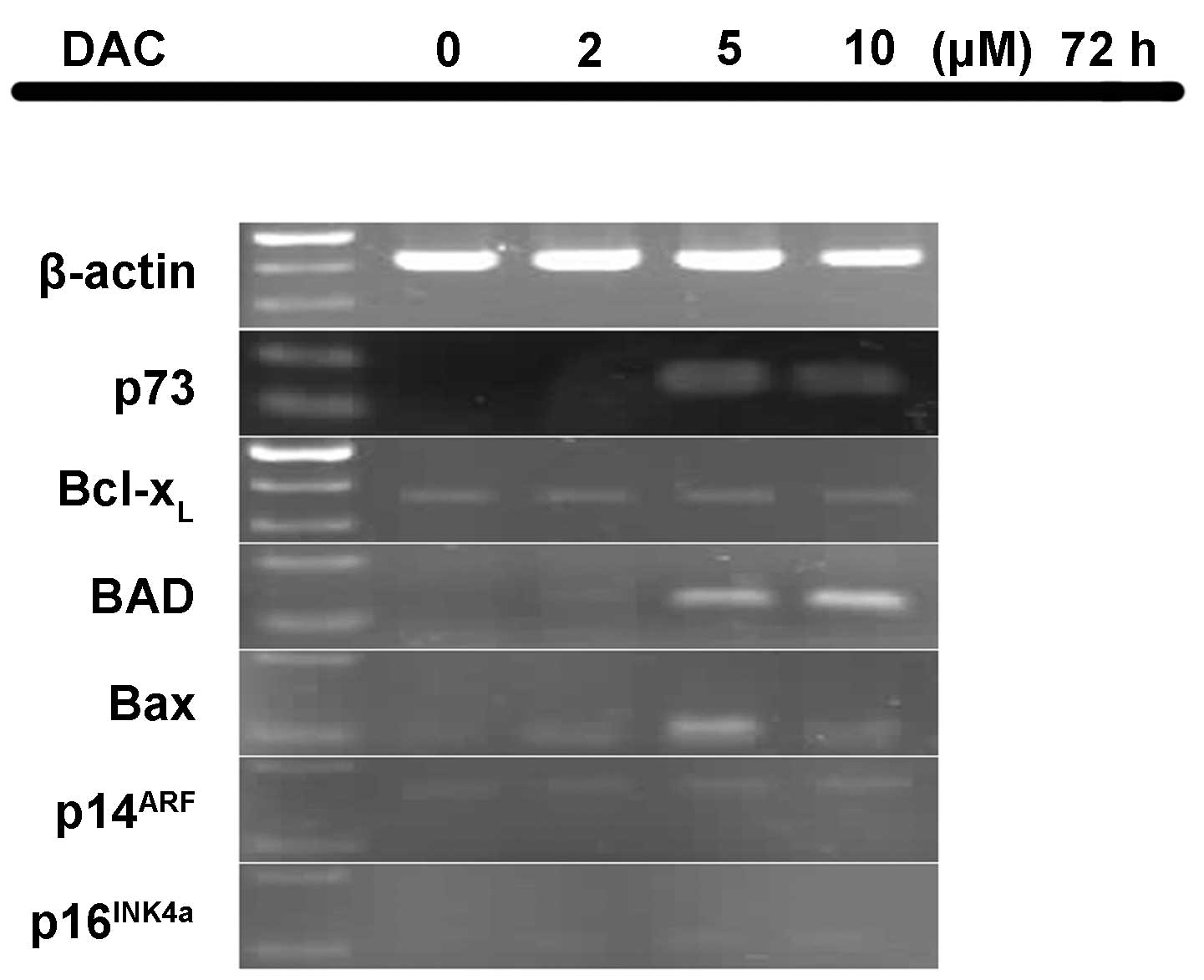
Effect of DAC on mRNA expression
levels
Following exposure to different concentrations of
DAC, the mRNA expression levels of six genes in the P15 cells was
determined (Fig. 3). The P15 cells
expressed no p73 mRNA following treatment with 0 and 2 µM
DAC; however, equal levels of p73 expression occurred upon
treatment with 5 and 10 µM DAC. Therefore, DAC appears to restore
the expression of p73 transcripts. In addition, no mRNA
expression of BAD was identified in the untreated P15 cells.
By contrast, treatment with 5 and 10 µM DAC markedly enhanced
BAD expression and treatment with 2 µM DAC weakly enhanced
BAD expression. In addition, p16INK4a was
expressed weakly upon treatment with 2, 5 and 10 µM DAC, while
Bax mRNA expression was evident at 5 µM DAC, and weakly
detected at 2 and 10 µM DAC. However, the mRNA expression levels of
p14ARF and Bcl-xL did not
appear to be affected by DAC treatment.
Cell apoptosis
Apoptotic cells are illustrated by fluorescence in
Fig. 4. A small number of apoptotic
cells were identified in the untreated cells or the cells treated
with DAC alone, with no statistically significant difference in the
mean number of apoptotic cells observed between these two groups.
The highest mean number of apoptotic cells was observed in the DAC
and CDDP combination therapy group (P=0.001, vs. control
group).
Discussion
Previous studies have indicated that methylation of
specific genes resulting in epigenetic silencing occurs in lung
cancer. These genes are involved in a number of processes,
including cell cycle control, DNA repair, the regulation of cell
differentiation and proliferation, and pro-apoptosis (13,14).
Therefore, the DNA methylation status of these genes may be used as
a molecular marker for early diagnosis, prognosis, disease
recurrence risk assessment or response to therapy in patients with
lung cancer (15,16).
The clinical efficacy of DAC therapy has yet to be
fully investigated in solid tumors; however, the combination of DAC
and a chemotherapeutic agent has been proven to exhibit a
synergistic effect. For instance, Charlet et al (17) reported that the treatment of
neuroblastoma cells with a combination of chemotherapeutic agents
and DAC significantly increased the levels of apoptosis induced by
CDDP, doxorubicin and etoposide therapy, when compared with
treatment with these chemotherapeutic agents alone. Furthermore,
Shang et al (18) demonstrated
that combination therapy with DAC and CDDP may be a novel strategy
to improve the clinical response rate of transitional cell
carcinoma of the bladder.
p73 is a member of the p53 family and, therefore,
p73 and p53 share significant homology in their DNA-binding
domains. Similar to p53, p73 can elicit cancer cell apoptosis in
response to DNA damage caused by CDDP-based chemotherapy. However,
the p73 gene is rarely mutated (19,20).
Alternatively, epigenetic silencing by promoter hypermethylation
and complex formation with inhibitory proteins are two distinct
inactivation mechanisms of p73 (21). Epigenetic silencing of the p73
gene via hypermethylation of its promoter was previously identified
in MDS and non-Hodgkin lymphoma (22,23).
Furthermore, the loss of p73 expression in six NSCLC cell
lines was determined to be associated with 5′CpG island
hypermethylation. In the C57 cell line, DAC was able to restore the
mRNA expression of p73 (24,25).
Similarly, the present study identified that DAC was successful in
restoring p73 mRNA expression in the human lung
adenocarcinoma cell line, P15 (Fig.
3). Furthermore, chemotherapeutic agent (CDDP)-induced cell
death was increased in the P15 cell line when administered in
combination with DAC (Fig. 4). Thus,
p73 appeared to enhance the chemosensitivity of P15 tumor
cells to CDDP, indicating a potential role of p73 as a tumor
suppressor in NSCLC.
p16INK4a, which is known to participate
in the regulation of the cell cycle, is frequently inactivated in a
variety of human cancer types. Kim et al (26) proposed that abnormal methylation of
p16INK4a was an early event of lung
carcinogenesis. Furthermore, hypermethylation of
p16INK4a was identified in plasma and sputum
samples of patients with early lung cancer, as well as in lung
cancer tissue samples (27,28). These studies indicated that
p16INK4a gene hypermethylation presented high
specificity as the hypermethylation did not occur in healthy lung
tissue. In addition, the abnormal methylation of
p16INK4a is associated with poor prognosis in
NSCLC patients (29) as the
disruption of p16INK4a cell cycle control would
allow clonal expansion to occur. In the present study, clone
formation and cell growth inhibitory assays demonstrated that the
tumor cell growth was significantly inhibited upon treatment with
DAC; this may indicate an association between restored expression
of p16INK4a and cell growth inhibition (Figs. 1, 2 and
3).
BAD and Bax are pro-apoptotic genes of
the Bcl-2 family; therefore, aberrant promoter methylation in these
genes may affect the tumor cell apoptosis pathway (30,31). In
the present study, the treatment of P15 cells with DAC increased
BAD and Bax mRNA expression levels (Fig. 3). Although the mechanisms involved
have yet to be fully elucidated, DAC may enhance pro-apoptosis in
P15 cells through the restoration of BAD and Bax
genes (Fig. 4).
In conclusion, the present study demonstrated that
the aberrant hypermethylation of various gene promoters affected
the chemosensitivity of human lung adenocarcinoma cells to
treatment with CDDP. Treatment of P15 cells with DAC restored the
mRNA expression levels of p73, p16INK4a,
BAD and Bax. Furthermore, combined therapy with DAC
and CDDP significantly suppressed the growth of lung tumor cells
compared with DAC or CDDP treatment alone, indicating the potential
of DAC in the treatment of NSCLC.
Acknowledgements
Data published in the present study were extracted
from the medical thesis of Mr. Wenyan Huang.
Glossary
Abbreviations
Abbreviations:
|
DAC
|
5-aza-2′deoxycytidine
|
|
CDDP
|
cisplatin
|
|
NSCLC
|
non-small-cell lung cancer
|
|
RT-PCR
|
reverse transcription-polymerase chain
reaction
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling
|
References
|
1
|
Field JK, Oudkerk M, Pedersen JH and Duffy
SW: Prospects for population screening and diagnosis of lung
cancer. Lancet. 382:732–741. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reck M, Heigener DF, Mok T, Soria JC and
Rabe KF: Management of non-small-cell lung cancer: recent
developments. Lancet. 382:709–719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ozawa Y, Inui N, Naitoh T, et al: Phase II
study of combination chemotherapy with S-1 and weekly cisplatin in
patients with previously untreated advanced non-small cell lung
cancer. Lung Cancer. 63:68–71. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barr MP, Gray SG, Hoffmann AC, et al:
Generation and characterisation of cisplatin-resistant non-small
cell lung cancer cell lines displaying a stem-like signature. PloS
One. 8:e541932013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sève P and Dumontet C: Chemoresistance in
non-small cell lung cancer. Curr Med Chem Anticancer Agents.
5:73–88. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Almeida GM, Duarte TL, Farmer PB, Steward
WP and Jones GD: Multiple end-point analysis reveals cisplatin
damage tolerance to be a chemoresistance mechanism in a NSCLC
model: implications for predictive testing. Int J Cancer.
122:1810–1819. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McCabe MT, Brandes JC and Vertino PM:
Cancer DNA methylation: molecular mechanisms and clinical
implications. Clin Cancer Res. 15:3927–3937. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheung HH, Lee TL, Rennert OM and Chan WY:
DNA methylation of cancer genome. Birth Defects Res C Embryo Today.
87:335–350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fang JY, Yang L, Zhu HY, Chen YX, Lu J, Lu
R, Cheng ZH and Xiao SD: 5-Aza-2′-deoxycitydine induces
demethylation and up-regulates transcription of p16INK4a gene in
human gastric cancer cell lines. Chin Med J (Engl). 117:99–103.
2004.PubMed/NCBI
|
|
10
|
Chu LC, Eberhart CG, Grossman SA and
Herman JG: Epigenetic silencing of multiple genes in primary CNS
lymphoma. Int J Cancer. 119:2487–2491. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Karahoca M and Momparler RL:
Pharmacokinetic and pharmacodynamic analysis of
5-aza-2′-deoxycytidine (decitabine) in the design of its
dose-schedule for cancer therapy. Clin Epigenetics. 5:32013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mund C, Hackanson B, Stresemann C, Lübbert
M and Lyko F: Characterization of DNA demethylation effects induced
by 5-aza-2′-deoxycytidine in patients with myelodysplastic
syndrome. Cancer Res. 65:7086–7090. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Belinsky SA: Gene-promoter
hypermethylation as a biomarker in lung cancer. Nat Rev Cancer.
4:707–717. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Belinsky SA: Silencing of genes by
promoter hypermethylation: key event in rodent and human lung
cancer. Carcinogenesis. 26:1481–1487. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pfeifer GP and Rauch TA: DNA methylation
patterns in lung carcinomas. Semin Cancer Biol. 19:181–187. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fleischhacker M, Dietrich D, Liebenberg V,
Field JK and Schmidt B: The role of DNA methylation as biomarkers
in the clinical management of lung cancer. Expert Rev Respi Med.
7:363–383. 2013. View Article : Google Scholar
|
|
17
|
Charlet J, Schnekenburger M, Brown KW and
Diederich M: DNA demethylation increases sensitivity of
neuroblastoma cells to chemotherapeutic drugs. Biochem Pharmacol.
83:858–865. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shang D, Liu Y, Matsui Y, et al:
Demethylating agent 5-aza-2′-deoxycytidine enhances susceptibility
of bladder transitional cell carcinoma to cisplatin. Urology.
71:1220–1225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Han S, Sermba S, Abe T, Makino N, Furukawa
T, Fukushige S, Takahashi H, Sakurada A, Sato M, Shiiba K, et al:
Infrequent somatic mutations of the p73 gene in various human
cancers. Eur J Surg Oncol. 25:194–198. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yoshikawa H, Nagashima M, Khan MA,
McMenamin MG, Hagiwara K and Harris CC: Mutational analysis of p73
and p53 in human cancer cell lines. Oncogene. 18:3415–3421. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maas AM, Bretz AC, Mack E and Stiewe T:
Targeting p73 in cancer. Cancer Lett. 332:229–236. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao Y, Fei C, Zhang X, et al: Methylation
of the p73 gene in patients with myelodysplastic syndromes:
correlations with apoptosis and prognosis. Tumour Biol. 34:165–172.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pei JH, Luo SQ, Zhong Y, Chen JH, Xiao HW
and Hu WX: The association between non-Hodgkin lymphoma and
methylation of p73. Tumour biol. 32:1133–1138. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu K, Zhan M and Zheng P: Loss of p73
expression in six non-small cell lung cancer cell lines is
associated with 5′CpG island methylation. Exp Mol Pathol. 84:59–63.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu K, Zhuang X and Mai Z: p73 expression
is associated with cellular chemosensitivity in human non-small
cell lung cancer cell lines. Oncol Lett. 5:583–587. 2013.PubMed/NCBI
|
|
26
|
Kim DH, Nelson HH, Wiencke JK, et al:
p16(INK4a) and histology-specific methylation of CpG islands by
exposure to tobacco smoke in non-small cell lung cancer. Cancer
Res. 61:3419–3424. 2001.PubMed/NCBI
|
|
27
|
Bearzatto A, Conte D, Frattini M, et al:
p16(INK4A) hypermethylation detected by fluorescent
methylation-specific PCR in plasmas from non-small cell lung
cancer. Clin Cancer Res. 8:3782–3787. 2002.PubMed/NCBI
|
|
28
|
Shin KC, Lee KH, Lee CH, Shin IH, Suh HS
and Jeon CH: MAGE A1-A6 RT-PCR and MAGE A3 and p16 methylation
analysis in induced sputum from patients with lung cancer and
non-malignant lung diseases. Oncol Rep. 27:911–916. 2012.PubMed/NCBI
|
|
29
|
Lou-Qian Z, Rong Y, Ming L, Xin Y, Feng J
and Lin X: The prognostic value of epigenetic silencing of p16 gene
in NSCLC patients: a systematic review and meta-analysis. PloS One.
8:e549702013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zinkel S, Gross A and Yang E: BCL2 family
in DNA damage and cell cycle control. Cell Death Differ.
13:1351–1359. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hervouet E, Vallette FM and Cartron PF:
Impact of the DNA methyltransferases expression on the methylation
status of apoptosis-associated genes in glioblastoma multiforme.
Cell Death Dis. 1:e82010. View Article : Google Scholar : PubMed/NCBI
|