Introduction
Pancreatoblastoma (PB) is a rare epithelial neoplasm
of the pancreas, typically occurring in the pediatric population.
Since the first report of PB in 1957, >200 cases have been
described in children, yet only 39 cases have been described in
adult patients (1,2).
PB is an aggressive and malignant tumour, exhibiting
high rates of local invasion, recurrence and distant metastatic
potential. Furthermore, adult patients with PB have a poor
prognosis compared with pediatric patients (3). Symptoms are usually vague and
radiological features are non-specific. Thus, diagnosis depends
largely on the identification of characteristic squamoid corpuscles
on histopathological examination, which would appear as whorled
nests of flattened cells with a squamous appearance.
Due to its rarity, no guidelines currently exist on
the management protocols for PB. Surgery is considered to be the
chief treatment strategy, while the role of chemoradiotherapy
remains unclear and the significant benefits of the currently
employed regimens are limited (3).
The current study presents the case of a 24-year-old
patient who was diagnosed with a rare pathology of PB, but
succumbed one year later due to disseminated metastatic disease. In
addition, a brief review of the relevant literature is discussed.
Written informed consent was obtained from the patient's
family.
Case report
In March 2013, a 24-year-old Caucasian, male patient
was admitted to the Department of Surgery, Konstantopouleio General
Hospital (Athens, Greece) with a three-week history of vague upper
abdominal discomfort, anorexia and weight loss. A physical
examination revealed obstructive jaundice and mild tenderness over
the epigastrium.
Laboratory tests identified elevated serum levels of
aspartate aminotransferase (247 IU/l; normal range, 10–37 IU/l),
alanine aminotransferase (289 IU/l; normal range, 12–78 IU/l),
alkaline phosphatase (563 IU/l; normal range, 46–116 IU/l),
γ-glutamyl transpeptidase (703 IU/l; normal range, 15–85 IU/l),
direct bilirubin (15.9 mg/dl; normal range, <0.3 mg/dl) and
total bilirubin (17 mg/dl; normal range, <1 mg/dl). However,
carcinoembryonic antigen (CEA), α-fetoprotein (AFP) and cancer
antigen (CA)19-9 levels were within the normal ranges.
An abdominal ultrasound demonstrated the presence of
an encapsulated and well-defined mass in the head of the pancreas.
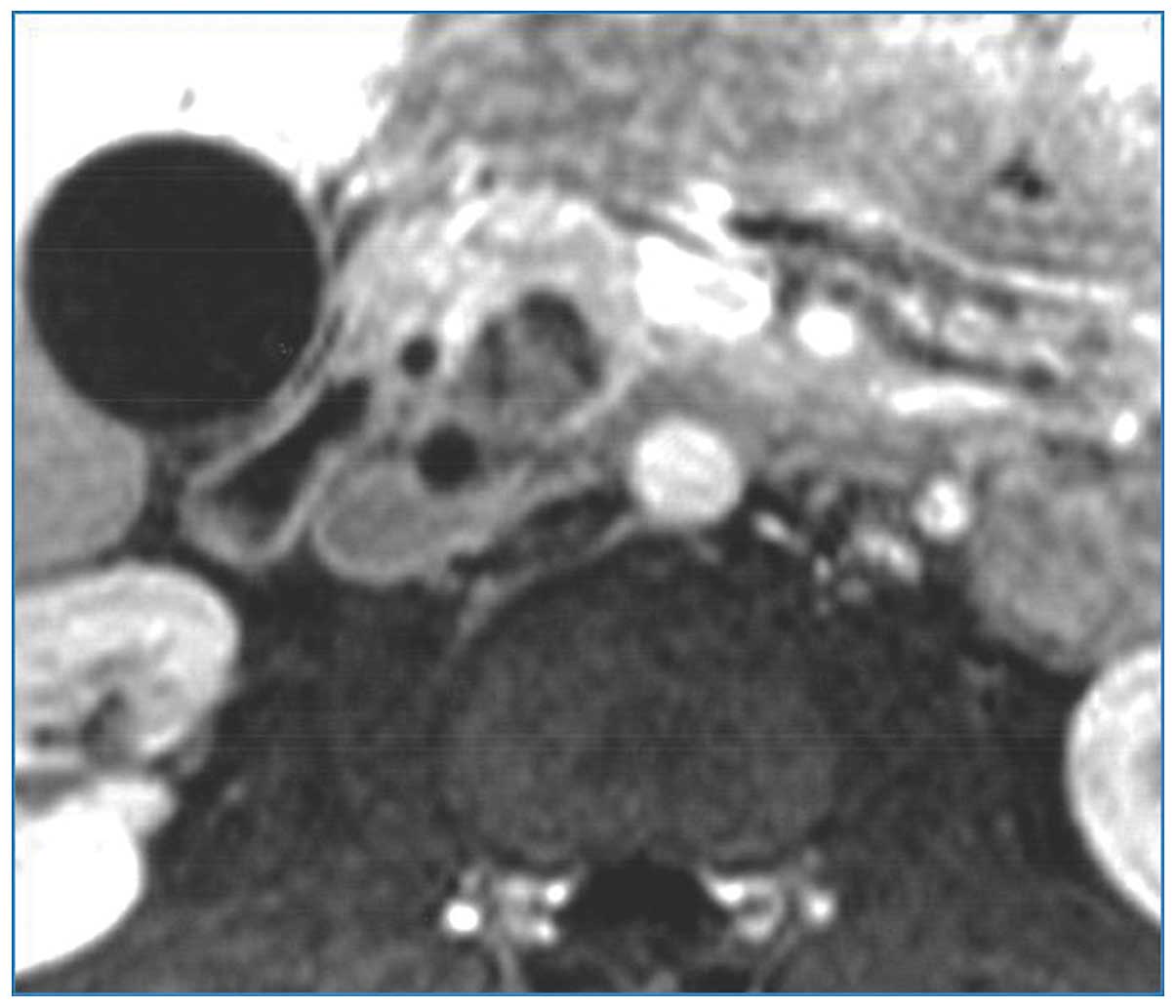
Contrast-enhanced computed tomography (CT) scans confirmed a
heterogeneous, hypodense mass in the pancreatic head, with no
dilatation of the main pancreatic duct and no vascular involvement
(Fig. 1). Furthermore,
contrast-enhanced magnetic resonance imaging indicated the presence
of a hypoenhancing, low-intensity mass in the posterior aspect of
the pancreatic head abutting the distal section of the common bile
duct on T1-weighted images (Fig. 2).
A high signal intensity was observed on T2-weighted images
(Fig. 3). In addition, magnetic
resonance cholangiopancreatography revealed obliteration of the
distal common bile duct, with marked proximal dilatation (Fig. 4).
The patient underwent an exploratory laparotomy,
during which a palpable mass measuring ~8×7×8 cm was identified in
the pancreatic head. No vascular invasion or distant metastases
were observed, therefore, a pylorus-preserving
pancreaticoduodenectomy was performed. The post-operative course
was uneventful and the patient was discharged on the seventh
post-operative day.
Histopathological analysis of the resected lesion
demonstrated typical features of PB. The tumour was composed of a
combination of undifferentiated small cells, and epithelial and
stromal components with partial encapsulation. The epithelial
component was dominant and demonstrated an acinar architecture,
solid sheets and squamoid corpuscles.
Follow-up abdominal CT scans were performed three
months post-operatively, and revealed multiple liver and bone
metastases. The patient underwent five courses of radiofrequency
ablation (15-minute courses of 60 W thermoablation with Elektrotom
106 HiTT needle, Berchtold GmbH and Co. KG, Tuttlingen, Germany)
combined with four cycles of systemic chemotherapy, consisting of
cisplatin (80 mg/m2 over 24 h) and doxorubicin (60
mg/m2 over 48 h), each course lasted three weeks;
however, no response was observed and the patient succumbed 13
months after initial diagnosis due to tumour dissemination.
Discussion
Adult PB is a rare neoplasm of epithelial origin,
accounting for <0.5% of exocrine pancreatic tumours (3). PB was initially described by Becker
(1) in 1957 as infantile pancreatic
carcinoma, however, Horie et al (4) coined the term pancreatoblastoma in 1977.
To date, since Palosaari et al (5) reported the first case of PB in a
37-year-old patient in 1986, only 39 cases have been published in
the literature (Table I) (3,5–33). A literature search of PubMed was
conducted with no language restrictions; studies published between
1986 and 2014 were searched using the key words
‘pancreatoblastoma’, ‘pancreas’, ‘pancreatic tumour’ and
‘pancreatic neoplasm’. Additional studies were identified from the
references of the retrieved papers. Cases of PB affecting patients
≥18 years of age were included in the analysis.
 | Table I.Adult pancreatoblastoma cases in the
literature (1986–2014). |
Table I.
Adult pancreatoblastoma cases in the
literature (1986–2014).
| First author/s
(ref.) | Year | Agea/gender | Symptoms | Tumour details | Treatment
strategy | Follow-up |
|---|
| Palosaari et
al (5) | 1986 | 37/M | Abdominal pain,
weight loss, diarrhea | 8 cm, head, LN and
vascular involvement | Incomplete resection,
chemoradiotherapy | With liver metastases
at 15 months |
| Hoorens et al
(6) | 1994 | 39/F | Abdominal mass | 13 cm, tail | Resection | NED at 30
months |
| Dunn and Longnecker
(7) | 1995 | 61/M | Splenomegaly | 9 cm, tail | Resection,
chemotherapy | Succumbed after 11
months |
| Klimstra et
al (8) | 1995 | 19/M | Abdominal mass | 15 cm, head; LN
involvement, multiple metastases | Resection | Succumbed after 10
months |
|
|
| 36/M | Obstructive
jaundice | ‘Large’, head, LN
involvement, liver metastasis | None | Succumbed after 5
months |
|
|
| 37/M | Abdominal mass,
weight loss | 12 cm, head, liver
metastasis |
Chemoradiotherapy | Succumbed after 38
months |
|
|
| 54/F | Abdominal pain | 20 cm, tail | Resection | NED at 15
months |
|
|
| 56/M | Abdominal mass | 20 cm, tail | Resection | NED at 5
months |
| Levey and Banner
(9) | 1996 | 68/F | Diarrhea, weight
loss | 9 cm, head | Resection | Succumbed after 4
months |
| Robin et al
(10) | 1997 | 20/M | Abdominal mass | 9 cm head | Resection,
chemotherapy | Succumbed after 7
months |
| Hayasaki et
al (11) | 1999 | 48/F | Urinary occult
blood | 5 cm, tail | Resection | NED at 15
months |
| Montemarano et
al (12) | 2000 | 20/F | Obstructive
jaundice | Head | Resection | No data |
| Mumme et al
(13) | 2001 | 22/F | Abdominal pain,
mass, weight loss | 9 cm, tail | Resection,
intraoperative radiotherapy, chemotherapy | Succumbed after 9
months |
| Benoist et
al (14) | 2001 | 48/F | Abdominal pain,
melaena | 10 cm, body, liver
metastasis | Resection,
metastasectomy, chemotherapy | NED at 36
months |
| Abraham et
al (15) | 2001 | 45/F | No data | No data | No data | No data |
|
| 2001 | 51/F | No data | No data | No data | No data |
| Gruppioni et
al (16) | 2002 | 30/M | Abdominal pain | 8 cm, head | Resection | NED at 10
months |
| Du et al
(17) | 2003 | 78/F | Obstructive
jaundice | 2.7 cm, ampulla of
Vater | Resection | NED at 6 months,
alive at 4 years |
| Pitman and Faquin
(18) | 2004 | 18/M | Abdominal pain,
weight loss, diarrhea | 9 cm, head,
vascular involvement | Neoadjuvant
chemoradiotherapy, resection, multiple metastasectomies | With lung
metastases at 7 years |
| Rosebrook et
al (19) | 2005 | 29/F | Abdominal pain | 2 cm, body | Resection | No data |
| Sheng et al
(20) | 2005 | 18/M | Abdominal pain,
obstructive jaundice | 10 cm, body | Resection, adjuvant
chemoradiotherapy, liver TAE | Succumbed after 26
months |
| Zhu et al
(21) | 2005 | 24/F | Obstructive
jaundice | 4 cm, body, liver
metastases | Chemotherapy,
unresectable | With liver
metastases at 9 months |
| Kuxhaus et
al (22) | 2005 | 69/M | Fever, weight
loss | ‘Large’, tail,
peritoneal carcinomatosis | Unresectable | No data |
| Rajpal et al
(23) | 2006 | 50/M | Abdominal pain,
weight loss | 13 cm, tail, colon
invasion, liver | Resection,
chemotherapy | Succumbed after 17
months metastasis |
| Charlton-Ouw et
al (26) | 2008 | 33/M | Abdominal pain,
mass, weight loss | 5 cm, head, liver
metastasis | Metastectomy,
resection, chemoradiotherapy | NED at 60
months |
| Ohike et al
(27) | 2008 | 74/F | Asymptomatic | 4.5 cm, head | Excision | NED at 108
months |
| Cavallini et
al (24) | 2009 | 69/M | Asymptomatic | 6 cm, body | Resection | NED at 15
months |
|
|
| 26/M | Abdominal pain | 5 cm, head | Resection | NED at 51
months |
| Comper et al
(25) | 2009 | 27/M | No data | 5.5 cm, head | Resection | No data |
|
|
| 69/M | No data | 5.5 cm, body | Resection | No data |
| Savastano et
al (28) | 2009 | 36/F | Obstructive
jaundice | 4.3 cm, head, LN
involvement | Resection, adjuvant
chemoradiotherapy | No data |
| Boix et al
(29) | 2010 | 33/F | Abdominal pain | 3.5 cm, body | Resection | Succumbed after 3
months |
| Balasundaram et
al (30) | 2012 | 27/F | Weight loss | 3.6 cm, body, liver
and lung metastases | Chemotherapy | Succumbed after 1
month |
| Gringeri et
al (31) | 2012 | 38/F | No data | Head | Resection,
chemotherapy, CyberKnife®, liver metastasectomies | NED at 44
months |
| Hammer and Owens
(32) | 2013 | 37/M | Abdominal pain,
obstructive jaundice | 7 cm, head | Resection | No data |
| Redelman et
al (33) | 2013 | 26/F | No data | 7.5 cm, head | Resection | No data |
| Salman et al
(3) | 2013 | 60/M | Abdominal pain | 1.8 cm, head, liver
metastasis | Resection,
chemotherapy | NED at 41
months |
|
|
| 51/M | Obstructive
jaundice, weight loss | 4 cm, head,
duodenum invasion, LN involvement | Chemotherapy,
resection | Succumbed after 51
months |
|
|
| 58/F | Abdominal pain | 4.5 cm, tail, LN
involvement | Resection | NED at 30
months |
| Present case | 2015 | 24/M | Abdominal pain,
weight loss, obstructive jaundice | 8 cm, head | Resection,
chemotherapy | Succumbed after 13
months |
PB displays a bimodal age distribution (modal ages,
2.5 and 40 years), while both genders are equally affected
(male:female ratio, 1:1) (3,31). The majority of cases are sporadic,
however, specific cases are associated with hereditary syndromes,
such as Beckwith-Wiedeman and familial adenomatous polyposis
syndromes (32).
Symptoms are typically vague and non-specific
(3,17,24,30). For
example, the majority of adult patients present with abdominal pain
or a palpable mass. Additionally, weight loss, anorexia and a
change in bowel habits are common symptoms on initial presentation.
PBs arising in the pancreatic head may cause biliary obstruction
and jaundice, as in the present case. Furthermore, patients may
manifest symptoms of endocrine abnormalities (15).
In the present literature review, the pancreatic
head, observed in 20/38 patients (53%) was the most common site of
tumour origin, followed by the tail, the body and the ampulla of
Vater (no tumour site data for two patients). The liver has been
found to be the most common site of distant metastasis and 25% of
cases are diagnosed with secondary liver disease upon initial
staging (19,24,30).
However, bone, pulmonary, peritoneal, brain and mediastinal
metastases have also been described (19,26).
In paediatric PB patients, elevated AFP and CEA
levels are the most common abnormal serological markers, and
elevated AFP expression has been reported in ≤68% of cases
(12,24,25,32), By
contrast, these PB tumour markers are typically within the normal
range in adult patients (1,3). Serum AFP levels may be used to monitor
clinical response to therapy or recurrence in those patients whose
tumors produce it (32).
Forming an accurate pre-operative diagnosis may be
difficult when based solely on radiographic features, as these are
non-specific and thus, PB cases may be mistaken for more common
entities, such as adenocarcinoma or neuroendocrine tumours. The
majority of PB tumours are well-defined, large and heterogeneous on
cross-sectional imaging, with mixed solid-cystic appearance
(3,19,24,26,30).
On magnetic resonance, the tumours are most commonly described as
heterogeneous with low-intermediate T1 signal intensity and high T2
intensity. Furthermore, PB tumours exhibit enhancement on
contrast-enhanced CT scans, with calcifications and internal
fibrous septa observed (3,24,30).
The differential diagnosis for PB includes benign
and malignant processes of the pancreas, including autoimmune
pancreatitis, adenocarcinoma, neuroendocrine tumours, solid
pseudopapillary tumours, mucinous cystic neoplasms and serous
microcystic adenoma (3,32).
Macroscopically, PB lesions are typically large (≤20
cm in diameter), partially-encapsulated, greyish or tan in colour,
with a soft consistency and areas of focal necrosis (3). Microscopic diagnosis may be difficult
due to histological heterogeneity. PB tumours often contain
multiple cell types, and demonstrate variable combinations of
ductal, acinar and neuroendocrine components. This variety of
elements is one of the distinctive pathological features of PB, the
other being the presence of squamoid corpuscles (i.e.,
circumscribed, whorled nests of flattened cells with a squamous
appearance, separated by dense stromal bands) (3,32).
Ductal adenocarcinoma, acinar cell carcinoma,
neuroendocrine neoplasms and solid pseudopapillary tumours all lack
the characteristic squamoid corpuscles observed in PB. Therefore,
pre-operative cytology is rarely useful, as accurate pre-operative
identification of squamoid corpuscles cannot be performed due to
sampling errors (3,18,21,33,34).
However, PB does not demonstrate the desmoplastic reaction that
occurs in adenocarcinoma, allowing differentiation from PB
(32).
Immunohistochemistry may facilitate the
characterisation of various components of the tumour. For example,
acinar cell lines stain positive for trypsin, chymotrypsin and
lipase, whereas neuroendocrine cells stain positive for
synaptophysin, chromogranin and neuron-specific enolase (3,32). In
addition, immunohistochemical analysis of AFP expression may be
positive within solid regions of the epithelial component.
Unlike pancreatic ductal adenocarcinoma, PB does not
appear to express the K-ras oncogene or p53 tumour suppressor
mutations (15,26). However, mutations of the adenomatous
polyposis coli/β-catenin pathway have been described, and allelic
loss on chromosome 11p (Wnt signaling pathway) is the most common
type of genetic alteration that has been identified in PB (12,15,26,35,36).
Furthermore, numerous paediatric cases of PB have occurred in
patients with Beckwith-Wiedemann syndrome and familial adenomatous
polyposis (12,15,19,26,32).
Due to the rarity of PB, an optimal treatment
strategy has yet to be standardised, however, in the adult
population, surgical resection remains the primary treatment
strategy (3,24). The roles of adjuvant chemotherapy and
radiotherapy remain under debate due to the small number of
patients treated thus far. Typically, chemotherapy with or without
radiotherapy has a role in the treatment of recurrent, residual,
unresectable and metastatic disease, although with variable
outcomes (3,24). However, the development of optimal
chemotherapeutic regimens remain under discussion due to the small
number of patients reported in the literature (26). In patients with incompletely resected
disease, post-operative radiotherapy may be administered as a
palliative treatment (3).
PBs exhibit malignant behaviour, with local
invasion, recurrence and distant metastasis, and adult PB patients
have a poorer prognosis compared with children, exhibiting
three-year survival rates of <40% (3,12,19,24).
Patients with unresected tumours have a median overall survival
time of five months, whereas surgery alone is associated with an
overall survival time of 15 months. Furthermore, treatment with
chemoradiotherapy following surgery appears to increase the overall
survival time to 20.5 months (3). The
limited number of reported cases does not allow for valid
conclusions to be drawn, however, positive lymph node involvement
has been associated with a poorer outcome (overall survival of 12
vs. 36 months with negative node involvement) (3). In addition, there is insufficient data
available to evaluate survival with or without vascular or
perineural invasion (3).
Although the benefits of neoadjuvant chemotherapy
have yet to be studied in randomised trials, it has demonstrated a
survival benefit in a small pediatric series (37). Of six pediatric patients treated with
neoadjuvant chemotherapy, five exhibited >50% tumour remission,
allowing for complete surgical resection in four of the five
patients, the fifth patient underwent a laparotomy, however the
tumour remained unresectable due to regional extension, and one
patient achieved complete tumour regression (37). Therefore, neoadjuvant chemotherapy may
have a role in the treatment of adult PB, predominantly in
identifying patients with responsive disease, who may be candidates
for surgical resection.
In conclusion, PB is an extremely rare neoplasm in
adults and the present study showcases its aggressive biological
and clinical behaviour. The patient presented with a three-week
history of obstructive jaundice and weight loss, and was diagnosed
with a pancreatic head mass. Following pancreaticoduodenectomy, the
patient developed multiple liver and bone metastases three months
later, and despite systemic chemotherapy and radiofrequency
ablation, succumbed to tumour dissemination. Pitfalls in the
pre-surgical diagnosis of PB, based currently on radiological and
cytological findings, and lack of management guidelines, due to the
paucity of the tumour, result in a generally unfavourable patient
prognosis.
Glossary
Abbreviations
Abbreviations:
|
PB
|
pancreatoblastoma
|
|
AFP
|
α-fetoprotein
|
|
CEA
|
carcinoembryonic antigen
|
References
|
1
|
Becker WF: Pancreatoduodenectomy for
carcinoma of the pancreas in an infant; report of a case. Ann Surg.
145:864–870; discussion. 870–872. 1957. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Argon A, Celık A, Onız H, Ozok G and
Barbet FY: Pancreatoblastoma, a rare childhood tumor: A case
report. Turk Patoloji Derg. 11:1–5. 2014.
|
|
3
|
Salman B, Brat G, Yoon YS, et al: The
diagnosis and surgical treatment of pancreatoblastoma in adults: a
case series and review of the literature. J Gastrointest Surg.
17:2153–2161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Horie A, Yano Y, Kotoo Y and Miwa A:
Morphogenesis of pancreatoblastoma, infantile carcinoma of the
pancreas: report of two cases. Cancer. 39:247–254. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Palosaari D, Clayton F and Seaman J:
Pancreatoblastoma in an adult. Arch Pathol Lab Med. 110:650–652.
1986.PubMed/NCBI
|
|
6
|
Hoorens A, Gebhard F, Kraft K, Lemoine NR
and Klöppel G: Pancreatoblastoma in an adult: its separation from
acinar cell carcinoma. Virchows Arch. 424:485–490. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dunn JL and Longnecker DS:
Pancreatoblastoma in an older adult. Arch Pathol Lab Med.
119:547–551. 1995.PubMed/NCBI
|
|
8
|
Klimstra DS, Wenig BM, Adair CF and
Heffess CS: Pancreatoblastoma. A clinicopathologic study and review
of the literature. Am J Surg Pathol. 19:1371–1389. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Levey JM and Banner BF: Adult
pancreatoblastoma: a case report and review of the literature. Am J
Gastroenterol. 91:1841–1844. 1996.PubMed/NCBI
|
|
10
|
Robin E, Terris B, Valverde A, Molas G,
Belghiti J, Bernades P and Ruszniewski P: Pancreatoblastoma in
adults. Gastroenterol Clin Biol. 21:880–883. 1997.(In French).
PubMed/NCBI
|
|
11
|
Hayasaki N, Miyake N, Takahashi H,
Nakamura E, Yamagishi S, Kuno Y, Mori N, Shinoda M, Kimura M,
Suzuki T and Tashiro K: A case of pancreatoblastoma in an adult.
Nihon Shokakibyo Gakkai Zasshi. 96:558–563. 1999.(In Japanese).
PubMed/NCBI
|
|
12
|
Montemarano H, Lonergan GJ, Bulas DI and
Selby DM: Pancreatoblastoma: imaging findings in 10 patients and
review of the literature. Radiology. 214:476–482. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mumme T, Büttner R, Peiper C and
Schumpelick V: Pancreatoblastoma: a rare malignant neoplasm in
early adulthood. Chirurg. 72:806–811. 2001.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Benoist S, Penna C, Julié C, Malafosse R,
Rougier P and Nordlinger B: Prolonged survival after resection of
pancreatoblastoma and synchronous liver metastases in an adult.
Hepatogastroenterology. 48:1340–1342. 2001.PubMed/NCBI
|
|
15
|
Abraham SC, Wu TT, Klimstra DS, Finn LS,
Lee JH, Yeo CJ, Cameron JL and Hruban RH: Distinctive molecular
genetic alterations in sporadic and familial adenomatous
polyposis-associated pancreatoblastomas: frequent alterations in
the APC/beta-catenin pathway and chromosome 11p. Am J Pathol.
159:1619–1627. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gruppioni F, Casadei R, Fusco F, Calculli
L, Marrano D and Gavelli G: Adult pancreatoblastoma. A case report.
Radiol Med. 103:119–122. 2002.PubMed/NCBI
|
|
17
|
Du E, Katz M, Weidner N, Yoder S, Moossa
AR and Shabaik A: Ampullary pancreatoblastoma in an elderly
patient: a case report and review of the literature. Arch Pathol
Lab Med. 127:1501–1505. 2003.PubMed/NCBI
|
|
18
|
Pitman MB and Faquin WC: The fine-needle
aspiration biopsy cytology of pancreatoblastoma. Diagn Cytopathol.
31:402–406. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rosebrook JL, Glickman JN and Mortele KJ:
Pancreatoblastoma in an adult woman: sonography, CT, and dynamic
gadolinium-enhanced MRI features. AJR Am J Roentgenol. 184:(Suppl).
S78–S81. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sheng L, Weixia Z, Longhai Y and Jinming
Y: Clinical and biologic analysis of pancreatoblastoma. Pancreas.
30:87–90. 2005.PubMed/NCBI
|
|
21
|
Zhu LC, Sidhu GS, Cassai ND and Yang GC:
Fine-needle aspiration cytology of pancreatoblastoma in a young
woman: report of a case and review of the literature. Diagn
Cytopathol. 33:258–262. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuxhaus L, Swayne LC, Chevinsky A and
Samli B: Adult metastatic pancreaticoblastoma detected with Tc-99m
MDP bone scan. Clin Nucl Med. 30:577–578. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rajpal S, Warren RS, Alexander M, et al:
Pancreatoblastoma in an adult: case report and review of the
literature. J Gastrointest Surg. 10:829–836. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cavallini A, Falconi M, Bortesi L, Crippa
S, Barugola G and Butturini G: Pancreatoblastoma in adults: a
review of the literature. Pancreatology. 9:73–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Comper F, Antonello D, Beghelli S, Gobbo
S, Montagna L, Pederzoli P, Chilosi M and Scarpa A: Expression
pattern of claudins 5 and 7 distinguishes solid-pseudopapillary
from pancreatoblastoma, acinar cell and endocrine tumors of the
pancreas. Am J Surg Pathol. 33:768–774. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Charlton-Ouw KM, Kaiser CL, Tong GX,
Allendorf JD and Chabot JA: Revisiting metastatic adult
pancreatoblastoma. A case and review of the literature. JOP.
9:733–738. 2008.PubMed/NCBI
|
|
27
|
Ohike N, Yamochi T, Shiokawa A, Yoshida T,
Yamazaki T, Date Y and Morohoshi T: A peculiar variant of
pancreatoblastoma in an adult. Pancreas. 36:320–322. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Savastano S, d'Amore ES, Zuccarotto D, et
al: Pancreatoblastoma in an adult patient. A case report. JOP.
10:192–195. 2009.PubMed/NCBI
|
|
29
|
Boix E, Yuste A, Meana A, Alcaraz E, Payá
A, Arnold C, Picó A and Lluis F: Corticotrophin-releasing
hormone-secreting pancreatoblastoma in an adult patient. Pancreas.
39:938–939. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Balasundaram C, Luthra M, Chavalitdhamrong
D, Chow J, Khan H and Endres PJ: Pancreatoblastoma: A rare tumor
still evolving in clinical presentation and histology. JOP.
13:301–303. 2012.PubMed/NCBI
|
|
31
|
Gringeri E, Polacco M, D'Amico FE, et al:
Liver autotransplantation for the treatment of unresectable hepatic
metastasis: an uncommon indication - a case report. Transplant
Proc. 44:1930–1933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hammer ST and Owens SR: Pancreatoblastoma:
a rare, adult pancreatic tumor with many faces. Arch Pathol Lab
Med. 137:1224–1226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Redelman M, Cramer HM and Wu HH:
Pancreatic fine-needle aspiration cytology in patients <35-years
of age: a retrospective review of 174 cases spanning a 17-year
period. Diagn Cytopathol. 42:297–301. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sigel CS and Klimstra DS: Cytomorphologic
and immunophenotypical features of acinar cell neoplasms of the
pancreas. Cancer Cytopathol. 121:459–470. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiao Y, Yonescu R, Offerhaus GJ, et al:
Whole-exome sequencing of pancreatic neoplasms with acinar
differentiation. J Pathol. 232:428–435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wood LD and Klimstra DS: Pathology and
genetics of pancreatic neoplasms with acinar differentiation. Semin
Diagn Pathol. 31:491–497. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Défachelles A, Martin De Lasalle E,
Boutard P, Nelken B, Schneider P and Patte C: Pancreatoblastoma in
childhood: clinical course and therapeutic management of seven
patients. Med Pediatr Oncol. 37:47–52. 2001. View Article : Google Scholar : PubMed/NCBI
|