Introduction
The ubiquitin-proteasome system (UPS) and
lysosome-dependent macroautophagy, also termed autophagy, are the
primary conserved intracellular pathways involved in protein
degradation. These pathways work together in order to maintain
homeostasis in eukaryotic cells (1).
UPS-mediated proteolysis affects several different proteins through
proteasome-mediated degradation, which is involved in the
regulation of the cell cycle, apoptosis and cellular
differentiation (2). Therefore,
targeting this pathway using proteasome inhibitors may represent a
novel approach for the treatment of cancer (3). A number of previous studies have
demonstrated that proteasome inhibitors can induce tumor cell death
via inhibiting proteasome activity (4,5).
Autophagy is an evolutionarily conserved
intracellular mechanism that degrades long-lived, misfolded
proteins and damaged organelles in order to maintain cellular
homeostasis by providing substrates and recycling amino acids and
nucleotides (6). Although autophagy
can be activated in a number of various cancer cells, including
esophageal cancer, by different approaches, such as
chemoradiotherapy (7), the exact role
of autophagy in cancer cells is complex. In certain circumstances,
autophagy demonstrates a protective role in cancer cells, whereas
in others, it is involved in type II programmed cell death, termed
autophagic cell death (8). One
previous study reported that when UPS is inhibited, autophagy is
upregulated (9). This suggests that
the UPS and autophagy may act as two compensatory mechanisms that
modulate protein degradation.
Esophageal carcinoma is the eighth most common cause
of cancer-associated mortality worldwide (10). Esophageal squamous cell carcinoma
(ESCC) is the main subtype in developing countries, particularly in
China (11). At the time of
diagnosis, a large proportion of patients with ESCC have lost the
optimum opportunity for surgery. The five-year survival rate for
ESCC remains extremely low as a result of resistance to anti-cancer
therapies, including chemotherapy and radiotherapy (12). Previous studies have indicated that
the inhibition of autophagy can potentiate
chemo-radiotherapy-induced apoptosis in ESCC cells (13,14). Due
to the fact that the proteasome inhibitor has become a novel target
for cancer therapy, the present study investigated whether
autophagy could be activated by the inhibition of the proteasome in
the ESCC cells. Furthermore, the ability of the inhibition of
autophagy to enhance proteasome inhibitor-induced ESCC cell death
was also investigated.
Materials and methods
Cell lines and culture
The poorly-differentiated ESCC EC9706 cell line was
purchased from the State Key Laboratory of Molecular Oncology,
Chinese Academy of Medical Sciences (Beijing, China). First, the
EC9706 cells were cultured in RPMI-1640 medium (Gibco Life
Technologies, Carlsbad, CA, USA) containing 10% fetal serum (FBS)
(GE Healthcare Life Sciences, Logan, UT, USA), 100 µg/ml
streptomycin and 100 units/ml penicillin in a humidified 5%
CO2 atmosphere at 37°C. The medium was changed every two
days. The cells were assigned to the control, MG-132 and MG-132
plus 3-methyladenine (3-MA) groups. Subsequent to a 24-h
incubation, the culture medium was replaced with fresh medium
containing no additional reagents for the control group, MG-132 for
the MG-132 group, or a combination of MG-132 and 3-MA for the
MG-132 plus 3-MA group. The EC9706 cells were then treated,
depending on the group, with 20 µM MG-132, 5 mM 3-MA or the two
combined for 24 h.
Chemical reagents
The MG-132 was purchased from EMD Millipore
(Billerica, MA, USA). First, the MG-132 was dissolved in
phosphate-buffered saline (PBS) to a storage concentration of 50
mM. Next, 3-MA (Sigma-Aldrich, St. Louis, MA, USA) was dissolved in
PBS to generate a 100 mM stock solution and was maintained at room
temperature until use. The subsequent dilution was made using
RPMI-1640 medium in order to create the desired concentration.
Monodansylcadaverine (MDC; Sigma-Aldrich) was used to assess
autophagy. The antibodies against Beclin-1 and caspase-9 were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA).
Measurement of cell viability and
apoptosis
Cell viability was detected using the cell counting
kit 8 (CCK-8) assay. First, the cells were seeded into a 96-well
flat bottom microplate at a density of 1×105 cells in
100 µl per well. Subsequent to treatment with MG-132, 3-MA or the
two combined, 100 µl medium was replaced with an equal volume of
fresh medium containing 10% CCK-8 (WST-8; Dojindo Laboratories,
Tokyo, Japan). Next, the cells were incubated for 3 h at 37°C, and
the absorbance of the solution was analyzed at 450 nm using a
microplate spectrophotometer (BioTek EL 340; BioTek Instruments,
Inc., Winooski, VT, USA). The cell viabilities were then calculated
using the following equation:
[TeX:] \begin{document}\begin{equation}
\text{Cell viability} (\%)=(1-{\mathrm{A}}_{450\
{\mathrm{sample}}}/{\mathrm{A}}_{450\ {\mathrm{control}}}) \times
100 \end{equation}\end{document}
Each experiment was performed in triplicate.
Cell apoptosis was detected by flow cytometry.
Subsequent to treatment, the attached and floating cells were
harvested and washed twice with PBS. Next, 5 µl Annexin
V-fluorescein isothiocyanate (FITC) and 5 µl propidium iodide (PI)
were added according to the instructions of the Annexin V-FITC
Apoptosis Detection kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing,
China). The cells were then incubated in the dark at room
temperature for 15 min. Finally, the cells were analyzed using a
flow cytometry system (FACScan; BD Biosciences, San Jose, CA, USA)
and the data were analyzed using Cell Quest software (BD
Biosciences).
Analysis of autophagy using MDC
In order to measure the autophagic ratio, the EC9706
cells were plated into 24-cell plates at a density of
1×105 cells. Subsequent to a 24-h incubation with the
different drugs, the cell pellets were suspended with 0.05 mM MDC
for 60 min at 37°C, washed with PBS three times, fixed with 4%
paraformaldehyde for 15 min at 4°C and then collected in 10 mM
Tris-HCl (pH 8.0) containing 0.1% Triton X-100. An inverted
fluorescence microscope (Olympus IX70; Olympus, Tokyo, Japan) was
then used to identify changes in the appearance of the autophagic
vacuoles and subsequently capture images. The fluorescence
intensity of the cells in the different groups was measured by flow
cytometry.
Western blotting
Subsequent to treatment with MG-132 alone or in
combination with the autophagic inhibitor 3-MA, the EC9706 cells
were harvested from cultured dishes and lysed in cold lysis buffer
for 20 min. The cell extracts were then collected and centrifuged
for 5 min at 9,180 × g. Overall, 20 µg total protein obtained from
the whole cell lysates were boiled in 1X SDS buffer for 5 min,
separated by 12% SDS-PAGE, and then electrotransferred using a
semi-dry transfer method to nitrocellulose membranes (GE Healthcare
Life Sciences, Uppsala, Sweden). Following electrophoretic
transfer, the membrane was blocked at 4°C overnight. Subsequent to
blocking, the membranes were incubated with the primary antibodies
at the recommended concentrations for 1 h. The The rabbit
polyclonal anti-human antibodies against caspase-3, Beclin-1, and
β-actin were obtained from Santa Cruz Biotechnology, Inc. Next, the
membranes were incubated with the anti-mouse immunoglobulin G
horseradish peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology, Inc.). Finally, the blots were developed using
enhanced chemiluminescence (GE Healthcare Life Sciences, Piscataway
NJ, USA) on Kodak X-omat LS film (Kodak, Rochester, NY, USA) and
the densitometry was performed using Kodak 1D Image Analyses
software, version 3.5 (Kodak).
Statistical analysis
All data represent at least three independent
experiments and are expressed as the mean ± standard deviation.
Student's t-test was used for the statistical analyses. P<0.05
was considered to indicate a statistically significant
difference.
Results
Proteasome inhibitor MG-132 inhibits
cell proliferation and induces cell death in ESCC cells
After the EC9706 cells were treated with various
concentrations of MG-132 or 3-MA for 48 h, the CCK-8 was used to
assess cell viability. As shown in Fig.
1, EC9706 cell growth was effectively inhibited by MG-132 or
3-MA in a dose- and time-dependent manner. The half maximal
inhibitor concentration of MG-132 at 48 h was 20±2.1 µmol/l.
Therefore, 20 µmol/l MG-132 was selected for further experiments.
In addition, it was revealed that MG-132 significantly inhibited
the proliferation of EC9706 cells. By contrast, the rates of cell
proliferation were significantly reduced in the MG-132 and 3-MA
combined treatment group.
3-MA reverses MG-132-induced autophagy
in the ESCC cell line
In order to determine the effect of proteasome
inhibition on autophagy, the fluorescence of MDC was used to
observe changes in the appearance of the autophagic vacuoles
(15). As shown in Fig. 2, there was no significant difference
in the autophagy vacuoles in the 3-MA-treated group compared with
the control group. However, the number of autophagic vacuoles
stained by MDC in the MG-132 group was markedly higher compared
with the control group. Following the addition of 3-MA, the number
of vacuoles was significantly decreased compared with the cells
treated with MG-132 alone. Furthermore, compared with the MG-132
group, the fluorescence intensity was decreased and the vacuoles
were reduced in the 3-MA and MG-132 combined group.
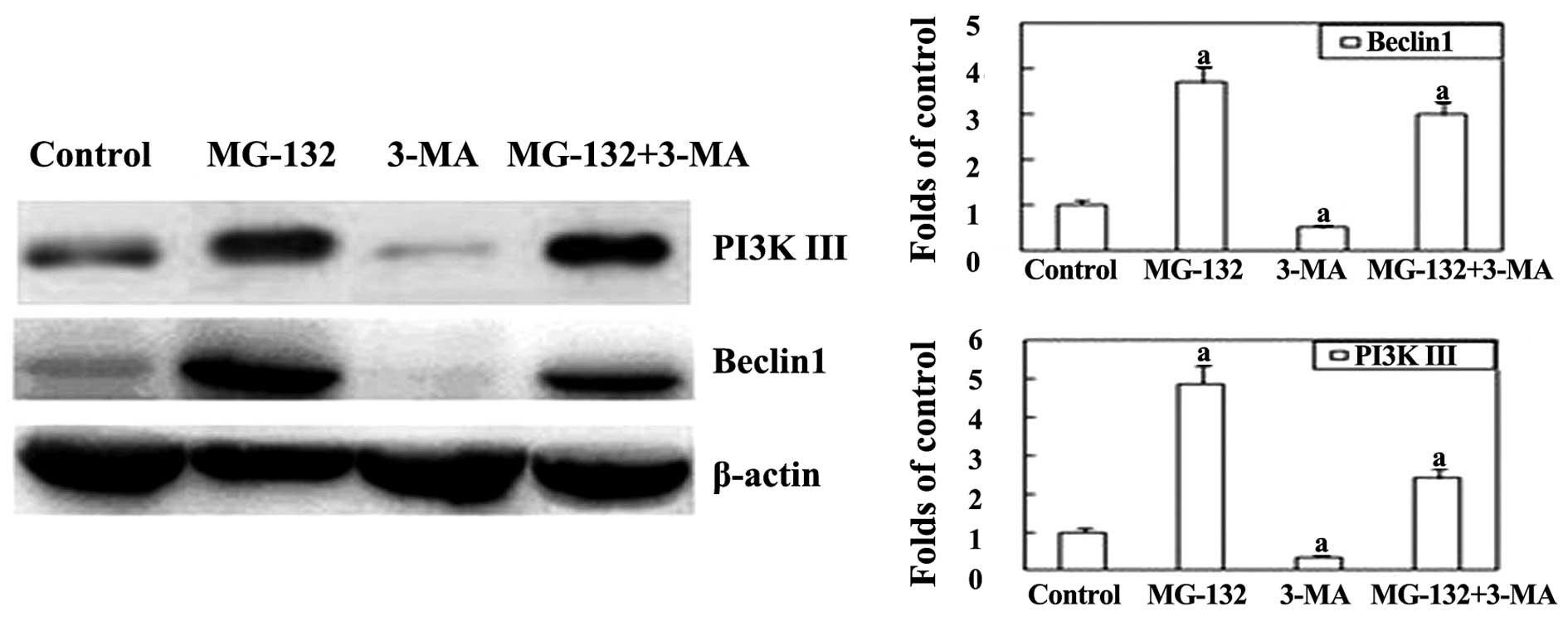
In order to investigate the underlying mechanism by
which the proteasome inhibitor MG-132 induces autophagy, western
blot analysis was performed. Beclin-1 is an important regulator
that promotes autophagy, and is also associated with a number of
biological processes, including development, immunity, adaptation
to stress, tumorigenesis, endocytosis, cytokinesis, aging and cell
death (16). As shown in Fig. 3, the expression of Beclin-1 was
significantly upregulated in the MG-132 group. However, following
the addition of 3-MA, the expression of Beclin-1 was markedly
inhibited. This indicates that autophagy induced by MG-132 can be
reversed by co-treatment with 3-MA in EC9706 cells.
MG-132-induced cell death increased by
3-MA in the ESCC cell line
CCK-8 assays were performed in order to investigate
the effect of autophagy inhibition on cell viability. As shown in
Fig. 4, 5 mmol/l 3-MA significantly
enhanced the inhibition of cell viability induced at 24 h by
MG-132. This suggests that autophagy inhibition can effectively
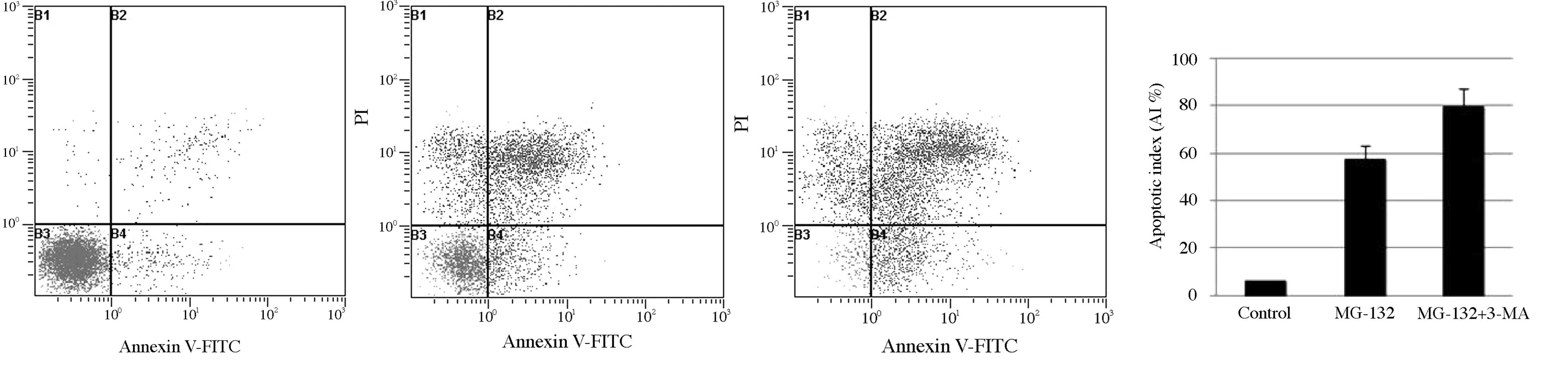
inhibit cell viability. In addition, the Annexin V-FITC and PI
staining assay was used to observe the apoptosis of ESCC cells
treated with MG-132. As shown in Fig.
4, the apoptosis rate of the EC9706 cells treated with MG-132
in combination with 3-MA significantly increased between 57.47 and
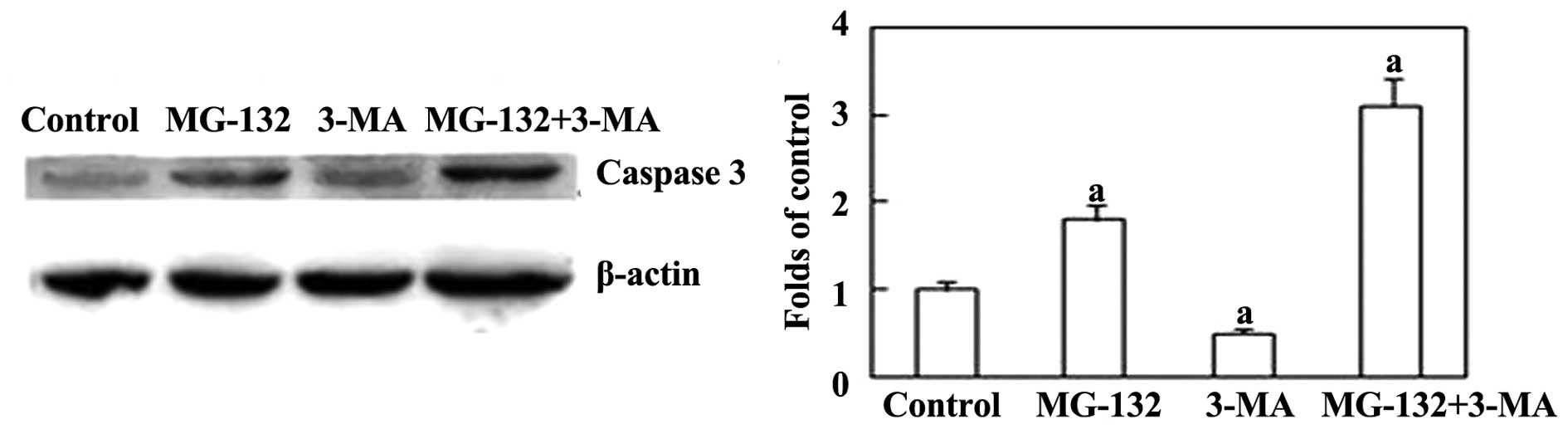
79.40% compared with the group treated with MG-132 alone. Next,
expression of the apoptosis-associated protein caspase-3 was
examined. Fig 5 shows that 3-MA
increased the levels of caspase-3 induced by MG-132. Together,
these results indicate that the inhibition of autophagy is able to
increase MG-132-induced apoptosis in ESCC cells.
Discussion
Chemotherapy is widely used for the treatment of
patients with metastatic or unresectable ESCC (17). However, ESCC cells have developed
resistance to chemotherapeutic drugs, which has resulted in a
reduction in the five-year survival rate. Therefore, a requirement
exists to identify novel therapeutic strategies or adjuvant drugs
for patients with ESCC. The present study revealed that inhibition
of the proteasome by MG-132 decreased cell proliferation, induced
cell death and activated autophagy in EC9706 cells. Furthermore,
the results demonstrated that MG-132 inhibited the proliferation in
EC9706 cells in dose- and time-dependent manner.
The proteasome system and autophagy machinery are
regarded as the two major cellular protein degradation systems
(3). It has been identified that
proteasome inhibitor-induced autophagy is able to control
endoplasmic reticulum stress and reduce cell death in cancer cells
by activating the downstream inositol-requiring enzyme-1/c-Jun
NH2-terminal kinase pathway (18). In
the present study, autophagy was assessed using biochemical
methods, including MDC and western blotting. As the results
demonstrate, the expression of the autophagy-associated protein,
Beclin-1, was significantly upregulated in EC9706 cells following a
48-h incubation with MG-132.
A number of studies have established that autophagy
can be activated in a variety of cancer cells under different
circumstances, including chemo-radiotherapy (9). However, the precise role of autophagy in
tumor cell death and survival remains unclear. In order to
investigate the role of autophagy in MG-132-induced EC9706 cell
apoptosis, the present study used the autophagy inhibitor, 3-MA, a
class III phosphatidylinositol 3-kinase inhibitor and a specific
inhibitor of autophagy. MG-132-induced autophagy in EC9706 cells
was significantly inhibited by the addition of 3-MA. Furthermore,
the inhibition of autophagy enhanced MG-132-induced apoptosis in
EC9706 cells through caspase-3 activation. This suggests that
caspase-3 may be the primary protease involved in the apoptosis
pathways (19). These findings
indicate that autophagy may be utilized as a protective mechanism
against cell death in MG-132-induced EC9706 cells, and that its
inhibition may enhance the EC9706 cell death induced by MG-132.
There are two important signaling pathways involved
in the process of autophagy, namely the PI3K/AKT/mTOR and class III
PI3K pathways. The specific inhibitor of autophagy, 3-MA, is known
to work by inhibiting the Beclin-1-PI3K III complex, which is a
component important for the formation of autophagosomes (20). The present study revealed that
following the addition of 3-MA, the expression of the PI3K III
protein was lower compared with that of the MG-132 group. These
results indicate that MG-132-induced autophagy in EC9706 cells may
be activated through the class III PI3K pathway.
In conclusion, the results of the present study
suggest that the proteasome inhibitor, MG-132, induces cell growth
inhibition and cell death in EC9706 cells. In addition, they
demonstrate that inhibition of the proteasome activates the process
of autophagy, and that MG-132-induced apoptosis is enhanced by
autophagy inhibition through the activation of the class III PI3K
pathway and the release of caspase-3. These findings suggest that
proteasome inhibitors may be potential novel anti-cancer agents for
the adjuvant treatment of ESCC.
Acknowledgements
This study was supported by the Youth Innovation
Fund Project of the First Affiliated Hospital of Zhengzhou
University and the Key Project of Science and Technology of the
Education Department of Henan province (grant no. 14A320076).
References
|
1
|
Wu WK, Cho CH, Lee CW, Wu K, Fan D, Yu J
and Sung JJ: Proteasome inhibition: a new therapeutic strategy to
cancer treatment. Cancer Lett. 293:15–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sorokin AV, Kim ER and Ovchinnikov LP:
Proteasome system of protein degradation and processing.
Biochemistry (Mosc). 74:1411–1142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wojcik S: Crosstalk between autophagy and
proteasome protein degradation systems: possible implications for
cancer therapy. Folia Histochem Cytobiol. 51:249–264. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Orlowski RZ and Kuhn DJ: Proteasome
inhibitors in cancer therapy: lessons from the first decade. Clin
Cancer Res. 14:1649–1657. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guo N and Peng Z: MG132, a proteasome
inhibitor, induces apoptosis in tumor cells. Asia Pac J Clin Oncol.
9:6–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu WK, Sakamoto KM, Milani M, et al:
Macroautophagy modulates cellular response to proteasome inhibitors
in cancer therapy. Drug Resist Updat. 13:87–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality and prevalence across five
continents: defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pisani P, Parkin DM, Bray F and Ferlay J:
Estimates of the worldwide mortality from 25 cancers in 1990. Int J
Cancer. 83:18–29. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu D, Yang Y, Liu Q and Wang J:
Inhibition of autophagy by 3-MA potentiates cisplatin-induced
apoptosis in esophageal squamous cell carcinoma cells. Med Oncol.
28:105–111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen YS, Song HX, Lu Y, et al: Autophagy
inhibition contributes to radiation sensitization of esophageal
squamous carcinoma cells. Dis Esophagus. 24:437–443. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Biderbik A, Kern HF and Elsässer HP:
Monodansylcadaverine (MDC) is a specific in vivo marker for
autophagic vacuoles. Eur J Cell Biol. 66:3–14. 1995.PubMed/NCBI
|
|
16
|
Wirawan E, Lippens S, Vanden Berghe T,
Romagnoli A, Fimia GM, Piacentini M and Vandenabeele P: Beclin1: a
role in membrane dynamics and beyond. Autophagy. 8:6–17. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Hagen P, Hulshof MC, van Lanschot JJ,
et al: CROSS Group: Preoperative chemoradiotherapy for esophageal
or junctional cancer. N Engl J Med. 366:2074–2084. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz
DB, Ron D and Yin XM: Linking of autophagy to ubiquitin-proteasome
system is important for the regulation of endoplasmic reticulum
stress and cell viability. Am J Pathol. 171:513–524. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harrington HA, Ho KL, Ghosh S and Tung KC:
Construction and analysis of a modular model of caspase activation
in apoptosis. Theor Biol Med Model. 5:262008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu YT, Tan HL, Shui G, et al: Dual role of
3-methyladenine in modulation of autophagy via different temporal
patterns of inhibition on class I and III phosphoinositide
3-kinase. J Biol Chem. 285:10850–10861. 2010. View Article : Google Scholar : PubMed/NCBI
|