Introduction
Gastric cancer accounts for a considerable amount of
global cancer mortality. It is most frequently detected in its
advanced stages. Peritoneal metastases in advanced gastric cancer
remain the leading reason for noncurative resection, recurrence
following surgery and poor outcomes (1). Type II cyclic guanosine monophosphate
(cGMP)-dependent protein kinase (PKG II), a serine/threonine
kinase, is an important regulator of diverse cellular processes.
Although classically recognized for its ability to modulate
intestinal secretion, bone growth and nervous system functions
(2–4),
PKG II has also been reported to act as an inhibitory component of
signal transduction processes in certain cancer cell types.
Swartling et al (5) found that
this kinase was involved in regulating cell proliferation in glioma
cells. Fallahian et al (6)
evaluated the significance of the downregulation of PKG II
expression in breast cancer and found that PKGII expression was
downregulated in the breast tumors compared to those of normal
tissue counterparts, which is an important evidence to support the
antitumor activity of this kinase. Previous results from our
laboratory demonstrated that PKG II inhibited the proliferation and
migration of gastric cancer cell lines through the suppression of
epidermal growth factor (EGF)-induced activation of the EGF
receptor and related signal transduction pathways (7,8).
Rho guanosine triphosphatases (GTPases) are members
of the small GTP-binding protein family that are involved in
diverse cell functions, including cytoskeletal organization,
migration, transformation, differentiation and proliferation
(8). Several members of this family
have been shown to be important in regulating cell migration: RhoA,
which stimulates the formation of actin stress fibers; Rac1, which
promotes the process of membrane ruffling; and Cdc42, which
enhances the extension of filopodia (9). Rho GTPases work as sensitive molecular
switches, existing in an inactive guanosine diphosphate (GDP)-bound
form or an active GTP-bound form. Activation of Rho GTPases is
regulated by guanine nucleotide dissociation inhibitors (GDIs),
guanine nucleotide exchange factors, and GTPase activating proteins
(10). Phosphorylation of Rho GTPases
is an additional mechanism by which the activity of these proteins
may be modulated, usually leading to their functional inactivation
(11,12). Our previous results indicated that PKG
II inhibits lysophosphatidic acid (LPA)-induced cell migration, by
phosphorylation of RhoA at Ser188, thereby decreasing its
activation (13).
The present study aimed to examine the potential
inhibitory effect of PKG II on Rac1 activity in LPA-induced cell
migration, and the underlying mechanism by which PKG II inhibits
Rac1 activation. Accumulating evidence indicates that
phosphorylation may be an important mechanism for the regulation of
Rac1 activation (12,14). Phosphorylation of Rac1 at Ser71 has
been reported to modulate downstream signaling by inhibiting the
interaction of Rac1 with its effectors (12). In addition,
phosphatidylinositol-4,5-bisphosphate 3-kinase/protein kinase B
(PI3K/Akt), mitogen-activated protein kinase kinase/extracellular
signal-regulated kinase (MEK/ERK) and phospholipase C γ1 (PLCγ1)
-mediated signal transduction have been implicated in the Rac1
signaling pathway in various human cancer cell lines (15–18).
Therefore, the current study also aimed to establish the effects of
inhibiting these pathways on Rac1 activation, and the ability of
PKG II to directly phosphorylate Rac1.
Materials and methods
Cell lines, plasmids and reagents
The human gastric cancer cell line AGS and human
embryonic kidney 293A cells were provided by the Institute of Cell
Biology (Shanghai, China). Adenoviral vectors encoding
β-galactosidase (pAd-LacZ) and PKG II (pAd-PKG II) were provided by
Dr. Gerry Boss and Dr. Renate Pilz of the University of California
(San Diego, CA, USA). The pEGFP-C2 vector containing dominant
negative Rac1 T17N insert was provided by Dr. Gu Luo of Nanjing
Medical University. Dulbecco's modified Eagle's medium (DMEM) and
fetal bovine serum (FBS) were purchased from Gibco (Grand Island,
NY, USA). The polyclonal rabbit anti-human PKG II was from Abgent
Biotechnology (San Diego, CA, USA, cat no. AP8001a) and the
application dilution was 1:200. The polyclonal rabbit anti-human
Rac1 was from Signalway Antibody LLC (College Park, MD, USA, cat
no. 21201) and the application dilution was 1:500. The polyclonal
rabbit anti human p-Rac1/Cdc42 (Ser71) and rabbit anti-human p-MEK
(Ser217/221) antibodies were from Cell Signaling Technology
(Danvers, MA, USA, cat no. 2461 and cat no. 9121S respectively) and
the application dilutions were all 1:1,000. The polyclonal rabbit
anti-human p-Ser/Thr antibody was from Abcam (Cambridge, MA, USA,
cat no. ab17464) and the application dilution was 1:1,000. The
polyclonal rabbit anti-human GFP antibody (cat no. BS6507),
polyclonal rabbit anti-human p-ERK (Thr202/Tyr204) antibody (cat
no. BS5016), polyclonal rabbit anti-human anti-p Akt (Ser473)
antibody (cat no. BS4006), and polyclonal rabbit anti-human p-PI3K
P85 (Tyr458)/P55 (Tyr199) antibody (cat no. BS4605) were purchased
from Bioworld Technology (St. Louis Park, MN, USA) and the
application dilutions were all 1:500. Horseradish peroxidase (HRP)
conjugated polyclonal goat anti-mouse IgG and polyclonal goat
anti-rabbit IgG secondary antibodies (cat no. 115-035-003 and
111-035-003, respectively) were from Jackson Immuno Research
Laboratories (West Grove, PA, USA) and the application dilutions
were all 1:10,000. The monoclonal mouse anti-Tag protein FLAG
antibody was used at a dilution of 1:2,000, LPA, U73122 and U0126
were from Sigma-Aldrich (St. Louis, MO, USA, cat no. F1804, cat no.
L7260, cat no. U6756 and cat no. U120 respectively). The cell
permeable cGMP analog 8-pCPT-cGMP was from Calbiochem (San Diego,
CA, USA). Mouse anti-FLAG, LPA, U73122 and U0126 were from
Sigma-Aldrich (St. Louis, MO, USA). LY294002 was from Beyotime
(Jiangsu, China). The cell transfection reagent Lipofectamin 2000,
PureLink RNA Mini kit and SuperScript III were from Invitrogen Life
Technologies (Carlsbad, CA, USA). Electrochemiluminescence (ECL)
reagents were from Millipore (Billerica, MA, USA). All other
reagents used were of analytical grade.
Preparation of adenoviral vectors
293A cells were transfected with pAd-LacZ or pAd-PKG
II, and cultured in DMEM, supplied with 10% FBS and maintained at
37°C in a humidified incubator with 95% air and 5% CO2
for up to 10 days until cytopathic effects were seen. The cells and
culture medium were subsequently harvested by pipetting up and down
gently in the growth medium until the cells were completely
resuspended. The cell suspension was transferred to a screw cap
centrifuge tube and the cells were pelletted by low speed
centrifugation (19) and then
underwent three freezing-thawing cycles. Ad-LacZ and Ad-PKG II were
amplified by using the supernatant containing these adenoviruses to
infect new 293A cells. The amplified adenoviral preparations were
titrated to determine the number of plaque forming units per ml,
and stored at −80°C until use.
Cell culture, transfection and
infection
AGS cells were cultured in DMEM supplemented with
10% FBS, and maintained at 37°C in a humidified atmosphere of 95%
air and 5% CO2. The medium was replaced every 2 days and
the cells were sub-cultured at confluence. For transfection with
plasmids, cells were sub-cultured the previous day, and
transfection was conducted according to the manufacturer's
instructions. The pEGFP-C2 was used as a control and the pRac1-T17N
and pFlag-Rac1 were used to express the dominant negative mutant
Rac1 and Flag-tagged Rac1, respectively. At 1 day prior to
infection with Ad-LacZ and Ad-PKG II, cells were freshly seeded at
70–80% confluence.
Cloning constructs
To generate FLAG-tagged wild type Rac1, total RNA
from AGS cells was isolated using a PureLink RNA Mini kit,
according to the manufacturer's instructions. The initial cDNA
strand was synthesized using SuperScript III, according to the
manufacturer's instructions. A fragment encoding the full-length
Rac1, with a HindIII site at the N terminus and a
BamHI site at the C terminus, was amplified by DNA by
polymerase chain reaction (PCR). The 5′ primer for wild type Rac1
was 5′-CCC AAG CTT ATG CAG GCC ATC AAG TGT GTG-3′, and the 3′
primer was 5′-CGC GGA TCC CAA CAG CAG GCA TTT TCT CTT CC-3′. The
PCR was performed with 50 ng of genomic DNA, 0.2 µM of each primer,
and 1X PrimeSTAR Max DNA polymerase (cat no. DR045A, Takara,
Dalian, China). The PCR amplification was performed as follows:
initial denaturation at 94°C for 2 min, followed by 30 cycles of
denaturation at 94°C for 15 sec, annealing at 64°C for 5 sec and
elongation at 72°C for 30 sec and then a final elongation at 72°C
for 1 min. The PCR products were cleaved using
HindIII-BamHI, and the target fragment was cloned
into the expression vector p3XFlag-myc-CMV-24.
Transwell migration assay
The migration activity of AGS cells was detected
using a transwell system (BD BioCoat Control, 8.0 mm PET membrane,
24-well cell culture inserts; BD Biosciences, San Jose, CA, USA).
Following trypsinization, 5×104 cells were seeded into
the upper chamber in serum-free culture medium. For LPA treatment,
LPA (10 µM) was added to the culture medium. For cGMP treatment,
cells were incubated with 8-pCPT-cGMP (250 µM) for 1 h prior to the
addition of LPA. Migration of the cells to the bottom of the
membrane was induced by incubation with medium containing 10% FBS
in the lower chamber for 12 h at 37°C in a tissue culture
incubator. The cells remaining in the upper chamber were removed
with cotton swabs. Cells that had migrated to the lower side of the
membrane were fixed in 4% paraformaldehyde solution for 30 min,
stained by Giemsa solution (10 min incubation) and rinsed with
water. Stained cells were subsequently examined by light
microscopy. Migrated cells were counted in five randomly selected
fields per insert, and mean values were calculated. All experiments
were performed in triplicate for each migration condition.
Western blot analysis
Protein samples were subjected to SDS-PAGE (8–12%)
according to the molecular size of the target protein;
electrophoresis and membrane transfer was performed following the
manufacturer's protocol (Bio-Rad, Hercules, CA, USA). The membranes
were incubated with primary antibodies overnight at 4°C in
tris-buffered saline and Tween 20 (TBS-T; 0.1% Tween 20), and the
corresponding secondary antibodies were incubated for 1 h at RT in
TBS-T (0.1% Tween-20), with three washes following each incubation.
ECL reagents were used to detect positive bands on the membrane.
For densitometry analysis, digital images of the positive bands
were obtained with Chemidoc XRS and analyzed using Quantity One
(Bio-Rad). Results were presented as the ratio of target protein:
loading control.
In vitro pull-down assay
48 h after transfection or infection, cells at ~90%
confluence on 100 mm culture plates were washed three times with
cold PBS and harvested in lysis buffer (25 mM HEPES pH 7.5, 150 mM
NaCl, 1% NP40, 10% glycerol, 25 mM NaF, 10 mM MgCl2, 0.25% sodium
deoxycholate, 1 mM EDTA, 1 mM Na3VO4, 10
mg/ml aprotinin, and 10 mg/ml leupeptin). For LPA treatment, 1µM
LPA was added to the culture medium for 1 min prior to cell lysis.
For cGMP treatment, cells were incubated with 8-pCPT-cGMP (250 µM)
for 1 h prior to LPA treatment. The supernatant obtained by
centrifugation (13,800 × g, 10 min) was subsequently incubated with
GST-Pak1 protein binding domain (GST-PBD; provided by Dr. Keith
Burridge in University of North Carolina, Chapel Hill, NC, USA) for
1 h at 4°C with agitation. Following thorough rinsing with lysis
buffer, the bound proteins were solubilized in 2X SDS sample buffer
and analyzed by western blotting with an anti-Rac1 antibody. To
indicate the amount of protein used in each pull-down assay, 5% of
the input lysate was loaded in the input lane. Densitometry
analysis was performed to quantify the positive bands, and the raw
volume ratio of active Rac1 to total Rac1 was calculated.
Immunoprecipitation
AGS cells grown on 100 mm culture plate were
transfected with FLAG-tagged plasmids containing wild type Rac1. At
6 h after transfection, the cells were infected by Ad-LacZ or
Ad-PKG II. Following a 48 h incubation, cells were treated with
8-pCPT-cGMP (250 µM) for 1 h before washing twice with cold PBS and
lysing with RIPA buffer (50 mM Tris-HCl pH 7.4, 1% Triton X-100, 1
mM EDTA, 1 mM leupeptin, 1 mM phenylmethylsulfonyl fluoride, 10 mM
NaF, 1 mM Na3VO4) 48 h following the
transfection and/or infection process. The supernatant was obtained
by centrifugation (13,800 × g, 10 min) and incubated with target
antibodies for 12 h at 4°C with agitation. Fresh protein G
conjugated to agarose was subsequently added, followed by a further
2 to 3 h incubation at 4°C with agitation. Immunoprecipitates were
centrifuged at 400 × g for 1 min at 4°C. The supernatant was
discarded and the pellet, which consisted of protein G conjugated
to agarose and Flag-tagged Rac1 protein, was washed four times with
binding buffer, prior to resuspension in an equal volume of 2xSDS
sample buffer. The precipitates were subsequently probed with
antibodies against the target proteins.
Statistical analysis
The data are presented as the mean ± standard
deviation (SD). Significance was assessed using analysis of
variance with SPSS statistical software. P<0.05 was considered
to indicate a statistically significant difference.
Results
PKG II inhibits LPA-induced cell
migration, which requires Rac1 activation
To evaluate the role of Rac1 in LPA-induced
migration of gastric cancer cells, Rac1 activity in AGS cells was
reduced by transfection with a plasmid (pEGFP-C2) containing cDNA
of Rac1 T17N (a dominant negative mutant of Rac1), and cell
migration following LPA stimulation (10 µM for 12 h) was assessed.
Cells transfected with the empty vector exhibited significantly
increased migration activity following the addition of LPA compared
with controls. However, in cells transfected with the Rac1 T17N
expression vector, the stimulatory effect of LPA on cell migration
was significantly lower compared with those transfected with the
empty vector (Fig. 1A). Expression
levels of Rac1-T17N were verified using western blotting with
anti-GFP antibody (Fig. 1B). The
results indicated that the activation of Rac1 was essential for
LPA-stimulated migration of gastric cancer cells.
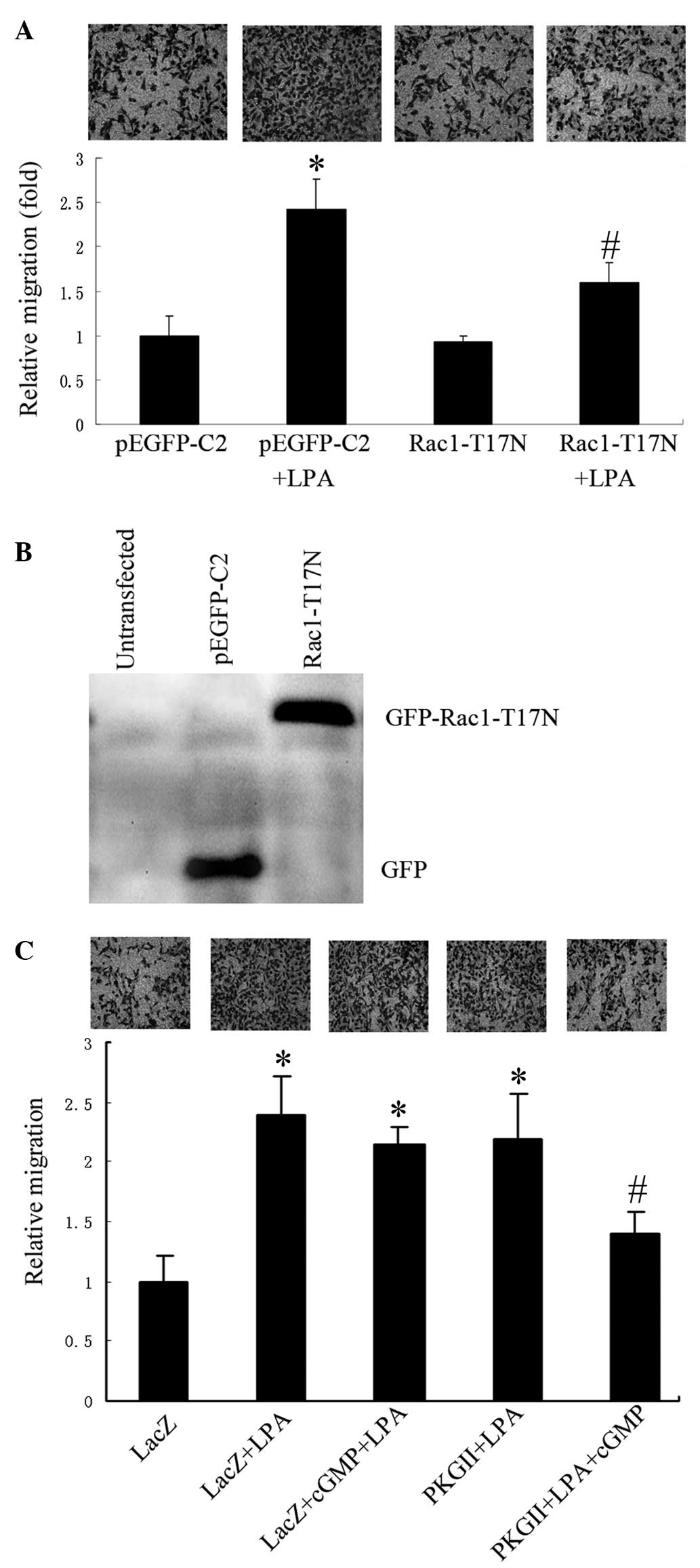 | Figure 1.PKG II inhibits LPA-induced cell
migration, which is dependent on Rac1 activation. The migration
activity of AGS cells was analyzed using transwell assays and cells
were examined under a light microscope (images above each graph;
magnification, ×200). (A) Relative migration of AGS cells
transfected with the empty pEGFP-C2 vector, or the vector
containing Rac1 T17N, in the presence or absence of LPA (mean ± SD
from three independent experiments performed in duplicate;
*P<0.01, compared with pEGFP-C2 group; #P<0.05,
compared with pEGFP-C2+LPA group). (B) Western blot analysis
showing expression of the inactive mutant Rac1-T17N in transfected
AGS cells, detected by anti-GFP antibody. (C) Relative migration of
AGS cells infected with either Ad-LacZ or Ad-PKG II, and treated
with LPA or LPA+8-pCPT-cGMP (mean + SD from three independent
experiments performed in duplicate; *P<0.01, compared with LacZ
group; #P<0.05, compared with LacZ+LPA group,
LacZ+cGMP+LPA group and PKG II+LPA group). LPA, lysophosphatidic
acid; GFP, green fluorescent protein; cGMP, cyclic guanosine
monophospate; PKG II, type II cGMP-dependent protein kinase; SD,
standard deviation. |
Subsequently, the effects of PKG II on LPA-induced
migration were investigated. AGS cells infected with Ad-LacZ or
Ad-PKG II were treated with 8-pCPT-cGMP (a cGMP analog for the
activation of PKG II) and/or LPA. A transwell migration assay
revealed that treatment with 8-pCPT-cGMP significantly inhibited
LPA-induced migration in the cells infected with Ad-PKG II, but not
in cells infected with Ad-LacZ (Fig.
1C)indicating that PKG II was capable of inhibiting LPA-induced
migration of AGS cells.
Pull-down assays confirmed that LPA induced Rac1
activation, and that the activity of Rac1 was markedly decreased
following treatment with 8-pCPT-cGMP in Ad-PKG II infected cells,
compared with that of Ad-LacZ infected cells (Fig. 2). The results indicate that inhibition
of LPA-induced migration by PKG II is associated with restriction
of Rac1 activity.
Phosphorylation is not associated with
PKG II inhibition on Rac1 activation
To examine the ability of PKG II to directly
phosphorylate Rac1 at Ser71, Rac1 was isolated by
immunoprecipitation, followed by western blotting with an
anti-p-Rac1/Cdc42 (Ser71) antibody. No difference in
phosphorylation on the Ser71 residue of Rac1 was observed following
8-pCPT-cGMP treatment in Ad-PKG II infected cells, compared with
that of Ad-LacZ infected cells (Fig.
3A). Following this, pan serine/threonine phosphorylation of
Rac1 was investigated in the immunoprecipitates. No increase in
phosphorylation of serine or threonine residues in Rac1 due to PKG
II was observed (Fig. 3B). These data
suggest that PKG II-mediated inhibition of Rac1 activation is not
due to direct phosphorylation of Rac1 by PKG II.
PKG II inhibition of Rac1 activity is
associated with activation of MEK/ERK and PI3K/Akt
Cells were pre-treated with LY294002 (20 µM), U0126
(10 µM) or U73122 (10 µM) to inhibit PI3K, ERK and PLCγ1,
respectively. Compared with LPA-treated cells in the absence of
pre-treatment, LPA-induced Rac1 activity was markedly reduced by
LY294002 pre-treatment, and partially reduced by U0126
pre-treatment (Fig. 4); pre-treatment
with U73122 had no effect on LPA-induced Rac1 activity. This
indicates that MEK/ERK and PI3K may act upstream of Rac1 in the
signaling pathway of LPA-induced migration.
The ability of PKG II to inhibit the activation of
the MEK/ERK and PI3K was also investigated. Western blotting
revealed that PKG II inhibited the LPA-induced
phosphorylation/activation of MEK and ERK (Fig. 5) and of PI3K and Akt (Fig. 6). These findings support the
conclusion that PKG II blocks Rac1 activation by inhibiting MEK/ERK
and PI3K/Akt pathways.
Discussion
PKG II is a subtype of PKG that has been implicated
in several physiological functions, including intestinal secretion,
bone growth, and nervous system activity (2–4).
Increasing evidence indicates that this enzyme may be important in
the activity of cancer cells; this may include inhibiting migration
and proliferation in certain cancer cell types (5,7,8). Our previous results demonstrated that
PKG II may inhibit LPA-induced migration by decreasing RhoA
activation (13). The results of the
current study suggest that PKG II may inhibit LPA-induced migration
of AGS cells by preventing Rac1 activation. Similar to RhoA, Rac1
is a member of the small Rho GTPases, which regulate important
cellular processes that are associated with cancer cell behavior,
such as migration (20). In
vivo and in vitro studies over the last decade have
firmly established the role of Rac1 in cancer cell invasion and
metastasis. For example, studies have demonstrated that Rac1 may
stimulate MMP-1 or MT1-MMP production in lung cancer cell lines and
enhance invasion in vitro (21,22). MMP-1
and MT1-MMP are matrix metalloproteinases (MMPs) belonging to a
family of extracellular matrix-degrading enzymes and are considered
to be important in cancer invasion and metastasis (21). The present study demonstrated that
Rac1 activation is essential for LPA-stimulated migration of AGS
cancer cells, providing further evidence for the importance of Rac1
in invasion and metastasis.
PKG signaling is known to inhibit RhoA activity
(23). However, whether Rac1
activation is increased or decreased by PKG II remains to be
determined (24–27). In the current study, the results of
the pull-down assay revealed that PKG II inhibited LPA-stimulated
activation of Rac1, and the direct and indirect effects of PKG II
on Rac1 activation were further investigated to determine the
mechanism by which this may occur. Protein kinases may directly
regulate the activity of Rho GTPases through phosphorylation, which
modulates the GDI affinity and the subcellular localization of the
GTPase, thereby negatively affecting its activity (28). RhoA may be phosphorylated by PKA/PKG
at Ser-188, and its phosphorylation status is associated with the
inhibition of its activity (11).
Recently, a similar mechanism was reported for Rac1: Thr108 is
phosphorylated by ERK in response to EGF, which alters the
interaction of Rac1 with PLC-γ1, affecting cell migration (29). Rac1 may also be phosphorylated by the
Akt kinase at Ser71, which results in reduced GTP-binding without
affecting GTPase activity (14,30).
Previous studies reported a PKG recognition site in Rac1,
suggesting that Rac1 may be directly phosphorylated by PKG II
(27); however, the results of the
present study found no evidence of this interaction.
Potential indirect mechanisms regulating Rac1
activity were also investigated. PI3K/Akt, MEK/ERK and
PLCγ1-dependent Ca2+ signaling pathways are major
signaling pathways in the regulation of LPA-related migration
(31–33). Hu et al (18) demonstrated that EGF stimulates
migration of hepatoma cell line HepG2 through GEP100-dependent
activation of the Arf6/ERK/Rac1 signaling pathway. Du et al
(16) reported that the PI3K and
ERK-induced Rac1 activation is required for hypoxia-induced HIF-1α
expression in breast cancer cells. Jones et al (15) proposed that the association of PLC-γ1
with complexes containing GIT1 and β-Pix is essential for its role
in integrin-mediated cell spreading and motility, during which the
activation of Cdc42 and Rac1, which are downstream of PLC-γ1, is
crucial since depletion of Cdc42 or Rac1 abolishes the elongated
cell phenotype. Therefore, the present study explored whether
MEK/ERK, PLC-γ1 or PI3K/Akt activation is essential in LPA-induced
Rac1 activation. The results revealed that specific chemical
inhibitors for PI3K and ERK caused a reduction in LPA-induced Rac1
activity. This indicated that LPA-induced Rac1 activation may be
mediated through PI3K and ERK pathways. The potential inhibitory
effect of PKG II on these signal pathways was also investigated,
revealing that PKG II prevented LPA-induced activation of PI3K and
ERK; this may be the mechanism by which PKG II inhibits gastric
cancer cell migration. However, it remains unclear how this
inhibition occurs. PKG may directly modify these proteins, or may
influence the upstream molecules of these pathways such as G
proteins or the LPA receptor.
In conclusion, the present study has demonstrated
that PKG II leads to the inhibition of LPA-induced activation of
Rac1 and associated migration in AGS cells. Preliminary
investigation of the mechanism indicates that PKG II does not
directly inhibit Rac1 through phosphorylation. However, it does
inhibit PI3K and ERK mediated signaling, which act upstream of
Rac1. Further studies are required to elucidate the specific
mechanisms.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81272755, 31040002 and
81201959) and the Specialized Research Fund for Senior Personnel
Program of Jiangsu University (grant no. 11JDG032). The authors
would like to thank Dr. Gerry Boss and Dr. Renate Pilz in the
University of California (San Diego, CA, USA) for the kind gifts of
adenoviral constructs.
References
|
1
|
Ferlay J, Soerjomataram II, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo MM, Parkin DM, Forman D and
Bray F: GLOBOCAN 2012 version 1.0, Cancer Incidence and Mortality
Worldwide: IARC CancerBase No. 11. International Agency for
Research on Cancer. [online]. 2013, https://gco.iarc.frAccessed. January
20–2014
|
|
2
|
Pfeifer A, Aszódi A, Seidler U, Ruth P,
Hofmann F and Fässler R: Intestinal secretory defects and dwarfism
in mice lacking cGMP-dependent protein kinase II. Science.
274:2082–2086. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miyazawa T, Ogawa Y, Chusho H, Yasoda A,
Tamura N, Komatsu Y, Pfeifer A, Hofmann F and Nakao K: Cyclic
GMP-dependent protein kinase II plays a critical role in C-type
natriuretic peptide-mediated endochondral ossification.
Endocrinology. 143:3604–3610. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tischkau SA, Mitchell JW, Pace LA, Barnes
JW, Barnes JA and Gillette MU: Protein kinase G type II is required
for night-to-day progression of the mammalian circadian clock.
Neuron. 43:539–549. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Swartling FJ, Ferletta M, Kastemar M,
Weiss WA and Westermark B: Cyclic GMP-dependent protein kinase II
inhibits cell proliferation, Sox9 expression and Akt
phosphorylation in human glioma cell lines. Oncogene. 28:3121–3131.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fallahian F, Karami-Tehrani F, Salami S
and Aghaei M: Cyclic GMP induced apoptosis via protein kinase G in
oestrogen receptor-positive and -negative breast cancer cell lines.
FEBS J. 278:3360–3369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen YC, Ren F, Sang JR, Tao Y and Xu WR:
Type II cGMP-dependent protein kinase inhibits proliferation of the
gastric cancer cell line BGC-823. Mol Med Rep. 3:361–366.
2010.PubMed/NCBI
|
|
8
|
Jiang L, Lan T, Chen Y, Sang J, Li Y, Wu
M, Tao Y, Wang Y, Qian H and Gu L: PKG II inhibits EGF/EGFR-induced
migration of gastric cancer cells. PLoS One. 8:e616742013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tapon N and Hall A: Rho, Rac and Cdc42
GTPases regulate the organization of the actin cytoskeleton. Curr
Opin Cell Biol. 9:86–92. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ellerbroek SM, Wennerberg K and Burridge
K: Serine phosphorylation negatively regulates RhoA in vivo. J Biol
Chem. 278:19023–19031. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schwarz J, Proff J, Hävemeier A, Ladwein
M, Rottner K, Barlag B, Pich A, Tatge H, Just I and Gerhard R:
Serine-71 phosphorylation of Rac1 modulates downstream signaling.
PLoS One. 7:e443582012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Chen Y, Li Y, Lan T and Qian H:
Type II cGMP-dependent protein kinase inhibits RhoA activation in
gastric cancer cells. Mol Med Rep. 9:1444–1452. 2014.PubMed/NCBI
|
|
14
|
Schoentaube J, Olling A, Tatge H, Just I
and Gerhard R: Serine-71 phosphorylation of Rac1/Cdc42 diminishes
the pathogenic effect of Clostridium difficile toxin A. Cell
Microbiol. 11:1816–1826. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jones NP and Katan M: Role of
phospholipase Cgamma1 in cell spreading requires association with a
beta-Pix/GIT1-containing complex, leading to activation of Cdc42
and Rac1. Mol Cell Biol. 27:5790–5805. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Du J, Xu R, Hu Z, Tian Y, Zhu Y, Gu L and
Zhou L: PI3K and ERK-induced Rac1 activation mediates
hypoxia-induced HIF-1α expression in MCF-7 breast cancer cells.
PLoS One. 6:e252132011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ray RM, Vaidya RJ and Johnson LR: MEK/ERK
regulates adherens junctions and migration through Rac1. Cell Motil
Cytoskeleton. 64:143–156. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu Z, Du J, Yang L, Zhu Y, Yang Y, Zheng
D, Someya A, Gu L and Lu X: GEP100/Arf6 is required for epidermal
growth factor-induced ERK/Rac1 signaling and cell migration in
human hepatoma HepG2 cells. PLoS One. 7:e387772012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schoofs G, Monica TJ, Ayala J, Horwitz J,
Montgomery T, Roth G and Castillo FJ: A high yielding serum-free,
suspension cell process to manufacture recombinant adenoviral
vectors for gene therapy. Cytotechnology. 28:81–89. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vega FM and Ridley AJ: Rho GTPases in
cancer cell biology. FEBS Lett. 582:2093–2101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ridley AJ: Rho proteins and cancer. Breast
Cancer Res Treat. 84:13–19. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soon LL, Yie TA, Shvarts A, Levine AJ, Su
F and Tchou-Wong KM: Overexpression of WISP-1 down-regulated
motility and invasion of lung cancer cells through inhibition of
Rac activation. J Biol Chem. 278:11465–11470. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Langer DA, Das A, Semela D, Kang-Decker N,
Hendrickson H, Bronk SF, Katusic ZS, Gores GJ and Shah VH: Nitric
oxide promotes caspase-independent hepatic stellate cell apoptosis
through the generation of reactive oxygen species. Hepatology.
47:1983–1993. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Muzaffar S, Shukla N, Bond M, Sala-Newby
G, Angelini GD, Newby AC and Jeremy JY: Acute inhibition of
superoxide formation and Rac1 activation by nitric oxide and
iloprost in human vascular smooth muscle cells in response to the
thromboxane A2 analogue, U46619. Prostaglandins Leukot Essent Fatty
Acids. 78:247–255. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Routray C, Liu C, Yaqoob U, Billadeau DD,
Bloch KD, Kaibuchi K, Shah VH and Kang N: Protein kinase G
signaling disrupts Rac1-dependent focal adhesion assembly in liver
specific pericytes. Am J Physiol Cell Physiol. 301:C66–C74. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rolli-Derkinderen M, Toumaniantz G, Pacaud
P and Loirand G: RhoA phosphorylation induces Rac1 release from
guanine dissociation inhibitor alpha and stimulation of vascular
smooth muscle cell migration. Mol Cell Biol. 30:4786–4796. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hou Y, Ye RD and Browning DD: Activation
of the small GTPase Rac1 by cGMP-dependent protein kinase. Cell
Signal. 16:1061–1069. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Forget MA, Desrosiers RR, Gingras D and
Béliveau R: Phosphorylation states of Cdc42 and RhoA regulate their
interactions with Rho GDP dissociation inhibitor and their
extraction from biological membranes. Biochem J. 361:243–254. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tong J, Li L, Ballermann B and Wang Z:
Phosphorylation of Rac1 T108 by extracellular signal-regulated
kinase in response to epidermal growth factor: a novel mechanism to
regulate Rac1 function. Mol Cell Biol. 33:4538–4551. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kwon T, Kwon DY, Chun J, Kim JH and Kang
SS: Akt protein kinase inhibits Rac1-GTP binding through
phosphorylation at serine 71 of Rac1. J Biol Chem. 275:423–428.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bian D, Su S, Mahanivong C, Cheng RK, Han
Q, Pan ZK, Sun P and Huang S: Lysophosphatidic Acid Stimulates
Ovarian Cancer Cell Migration via a Ras-MEK Kinase 1 Pathway.
Cancer Res. 64:4209–4217. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim EK, Yun SJ, Do KH, Kim MS, Cho M, Suh
DS, Kim CD, Kim JH, Birnbaum MJ and Bae SS: Lysophosphatidic acid
induces cell migration through the selective activation of Akt1.
Exp Mol Med. 40:445–452. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jans R, Mottram L, Johnson DL, Brown AM,
Sikkink S, Ross K and Reynolds NJ: Lysophosphatidic acid promotes
cell migration through STIM1- and Orai1-mediated Ca2+(i)
mobilization and NFAT2 activation. J Invest Dermatol. 133:793–802.
2013. View Article : Google Scholar : PubMed/NCBI
|



















