Introduction
Desmoplastic small round cell tumors (DSRCTs) are
uncommon and highly aggressive neoplastic entities. The
well-defined clinical and histological characteristics of DSRCT
were initially described in 1989 in a study by Gerald et al
(1) and then discussed again in 1991
in a study by Gerald and Rosai (2).
However, the associated clinical symptoms and radiological findings
are non-specific and similar to other primary intra-abdominal
neoplasms. Thus far, <200 cases of DSRCT have been reported in
the literature, with a higher incidence in children and young
adults, a male predominance (male : female ratio, 4:1), and an
average age of onset of 21 years (3).
Furthermore, patients typically present with vague abdominal
discomfort or distention.
The location of DSRCT is predominantly
intra-abdominal, exhibiting no clearly identifiable visceral
origin, but with diffuse peritoneal involvement and a clinical
presentation of pain, abdominal distention and abdominal masses
(2). Alternative primary sites,
including paratesticular, ovarian, lung, intracranial, thoracic,
and head and neck areas, have also been reported (4).
The most common sites of metastasis are the liver,
lymphoid tissue and peritoneum. Differentiation of DSRCT from other
small round cell tumors is important due to its highly aggressive
nature, with an average survival time of <2 years (2).
The reported 5-year survival rate is 15% (5), with an average survival time of 17
months (2); this prognosis is mainly
due to the lack of standardization in treatment, and the inadequate
response to radiation therapy and chemotherapy.
Various aggressive treatment regimens have been
applied for patients with DSRCT; however, no curative outcome or
notable impact on long-term survival has been noted (6).
The present study evaluated the response of two
patients with DSRCT to two distinct treatment strategies and
discussed the clinical findings of this tumor type. In addition, a
brief review of the relevant literature was performed.
Case report
Case 1
A 23-year-old male presented to the Clinic Hospital
of Botucatu School of Medicine (São Paulo State University, São
Paulo, Brazil) in September 2012 due to pain in the right side of
the abdomen and inguinal region, associated with difficulty
urinating and dyschezia that had been apparent for 3 months. The
patient was in a good clinical condition, with a palpable liver on
the right side and a mass in the right flank.
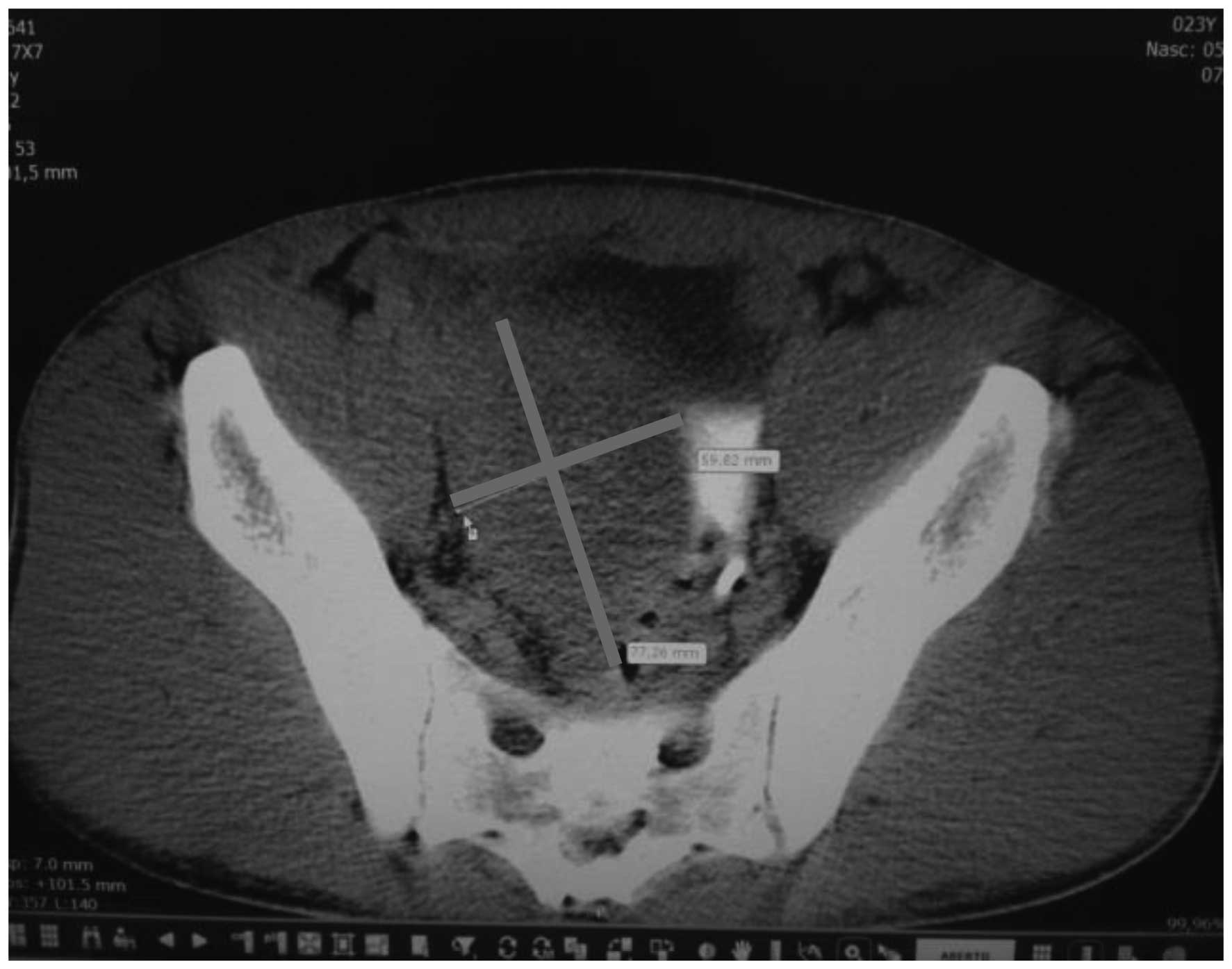
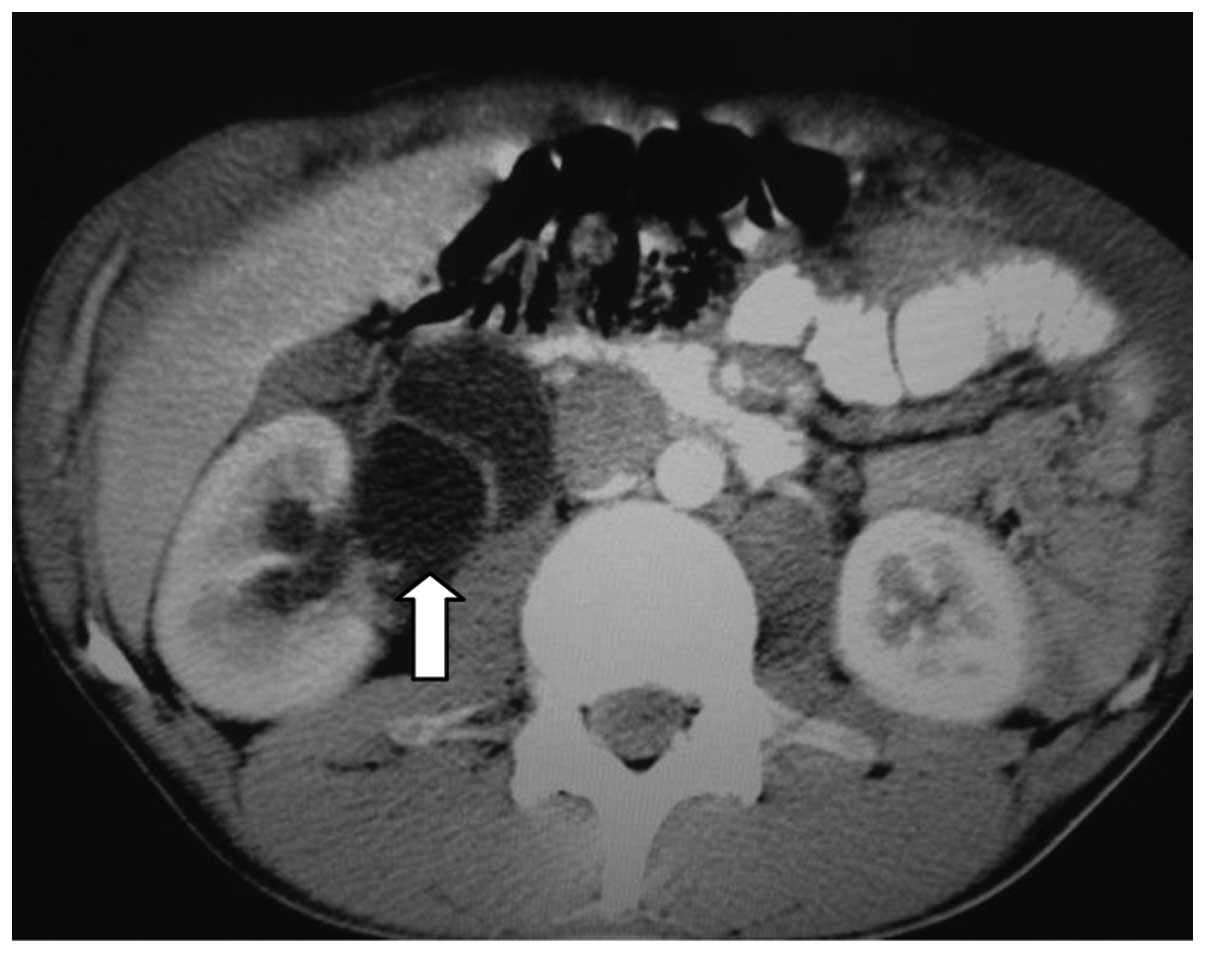
Computed tomography (CT) scans of the abdomen and
pelvis revealed a heterogeneous, hypovascular pelvic mass measuring
7.6×6.8 cm (Fig. 1). The mass was
located posteriorly and superiorly to the bladder, with thickening
of the rectum and a right large hydronephrosis (Fig. 2). Additionally, colonoscopy identified
extrinsic compression into the rectum. The differential diagnosis
was of a lymphoproliferative lesion or retroperitoneal sarcoma.
However, subsequent ultrasound-guided biopsy and histopathological
analysis of the pelvic mass indicated a morphology compatible with
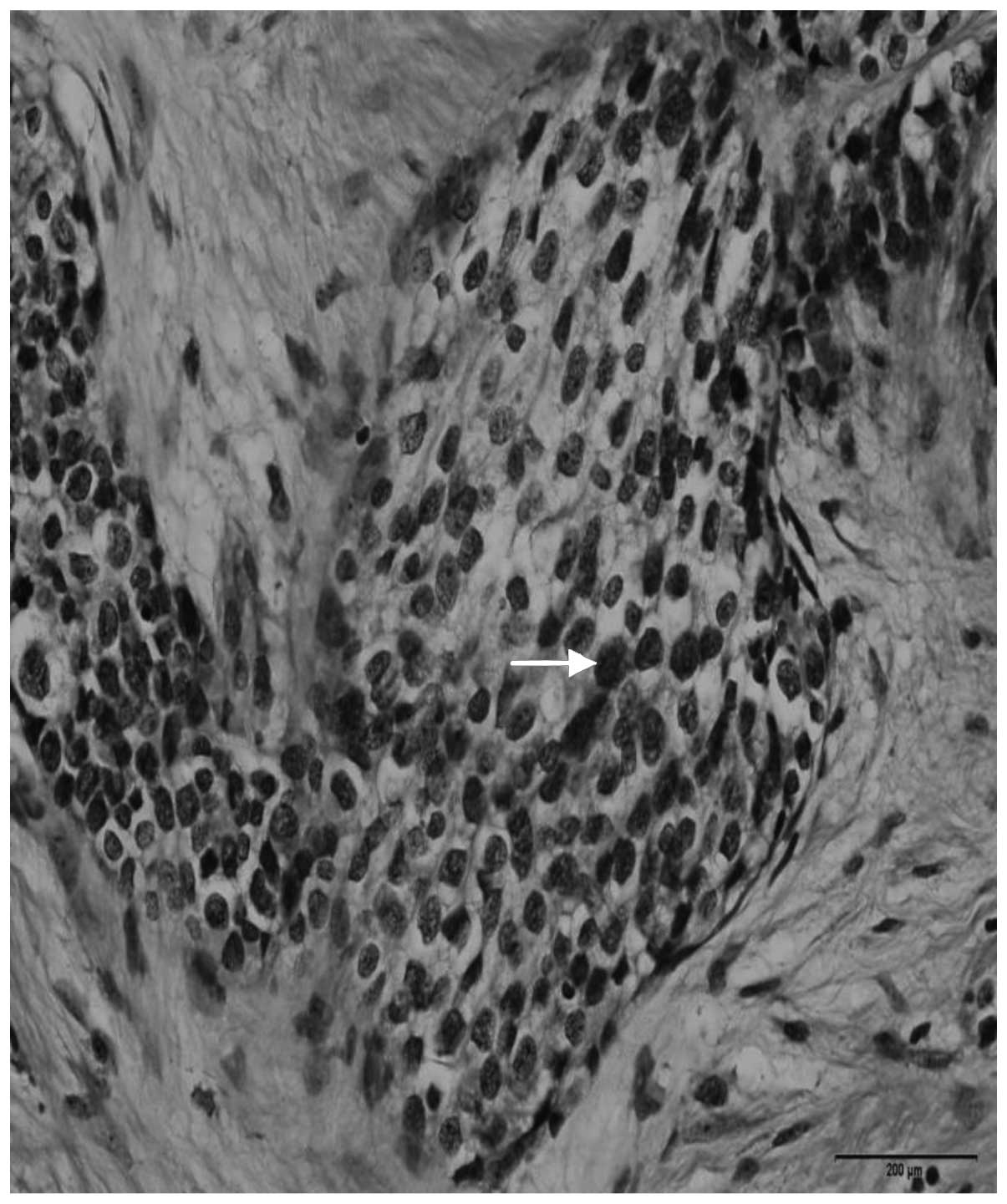
a high-grade malignant neoplasm. It was characterized by groups of
small cells featuring large and hyperchromatic nuclei with scant
cytoplasm, arranged in the desmoplastic stroma. Immunohistochemical
analysis of this sample revealed positivity for cytokeratin
(monoclonal mouse anti-human; clone, AE1/AE3) and desmin
(monoclonal mouse anti-human; clone, D33), and negativity for S100
protein (polyclonal rabbit anti-S-100), CD45 (leucocyte common
antigen; monoclonal mouse anti-human; clone, 2D1), myogenin
(monoclonal mouse anti-myogenin; clone, F5D), chromogranin
(polyclonal rabbit anti-human) and WT-1 (Wilms' tumor suppressor
gene 1; monoclonal mouse anti-human; clone, 6FH2; all purchased
from Dako North America, Inc., Carpinteria, CA, USA). The CD45
negativity excluded a diagnosis of lymphoma. These morphological
and immunohistochemical findings indicated a diagnosis of DSRCT
(World Health Organization classification, 2013) (7).
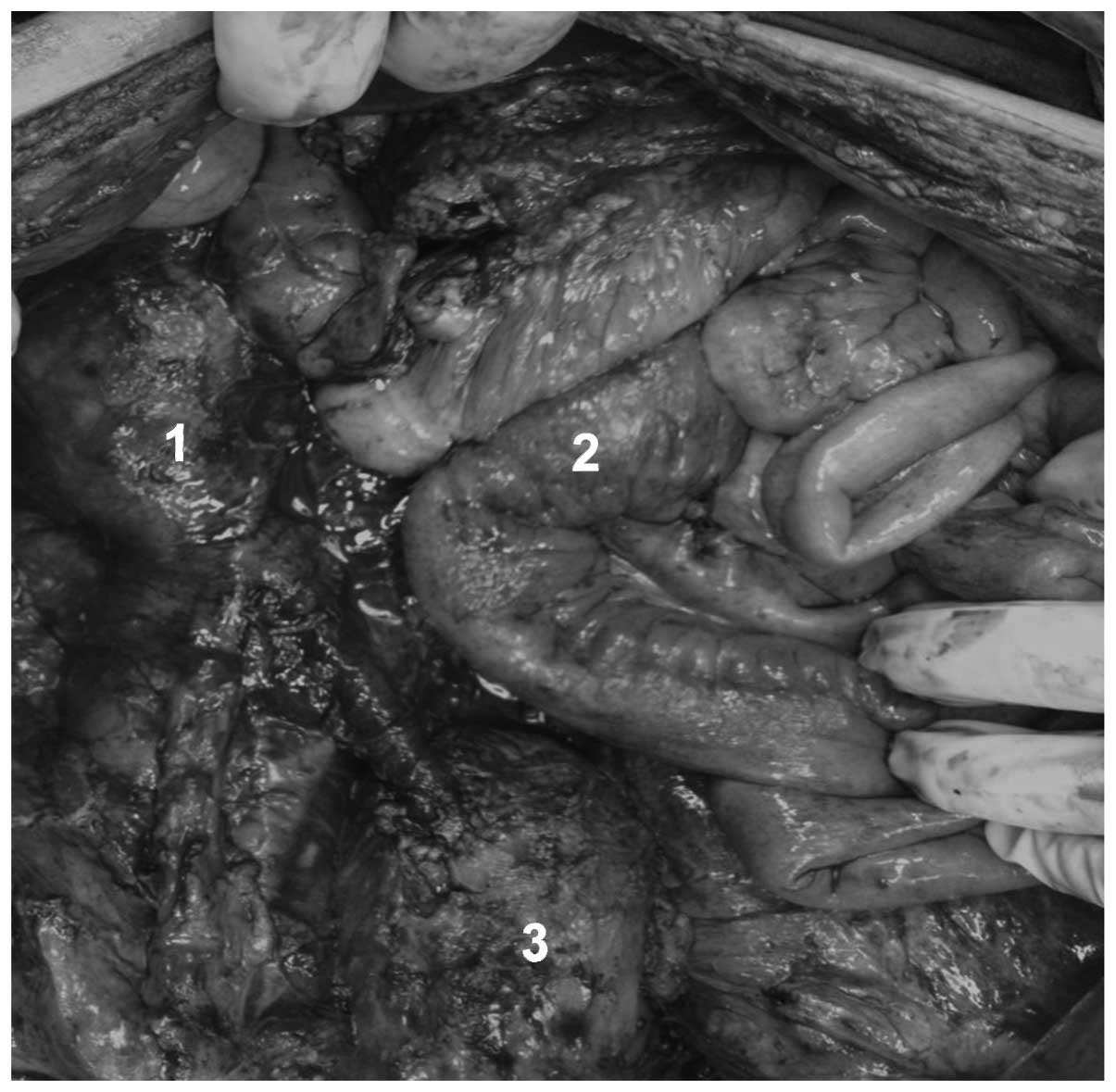
During a laparotomy, right hydronephrosis was
observed that was caused by a large tumor involving the cecum,
terminal ileum and right ureter. Implantation of the tumor was
identified in the right colon, liver and pelvic cavity, with
involvement of the rectum. Consequently, a resection of the
terminal ileum, cecum, right colon, distal segment of ureter,
sigmoid colon and middle rectum was performed. In addition, a
right, left and pelvic peritoniectomy was performed. Intestinal
reconstruction was re-established with an ileo-transverse
anastomosis associated with a left colostomy, implantation of a
proximal urether into the bladder and insertion of a double-J
catheter (Figs. 3 and 4). The post-operative follow-up was
uneventful, however, deep vein thrombosis occurred in the right
lower limb 20 days after surgery.
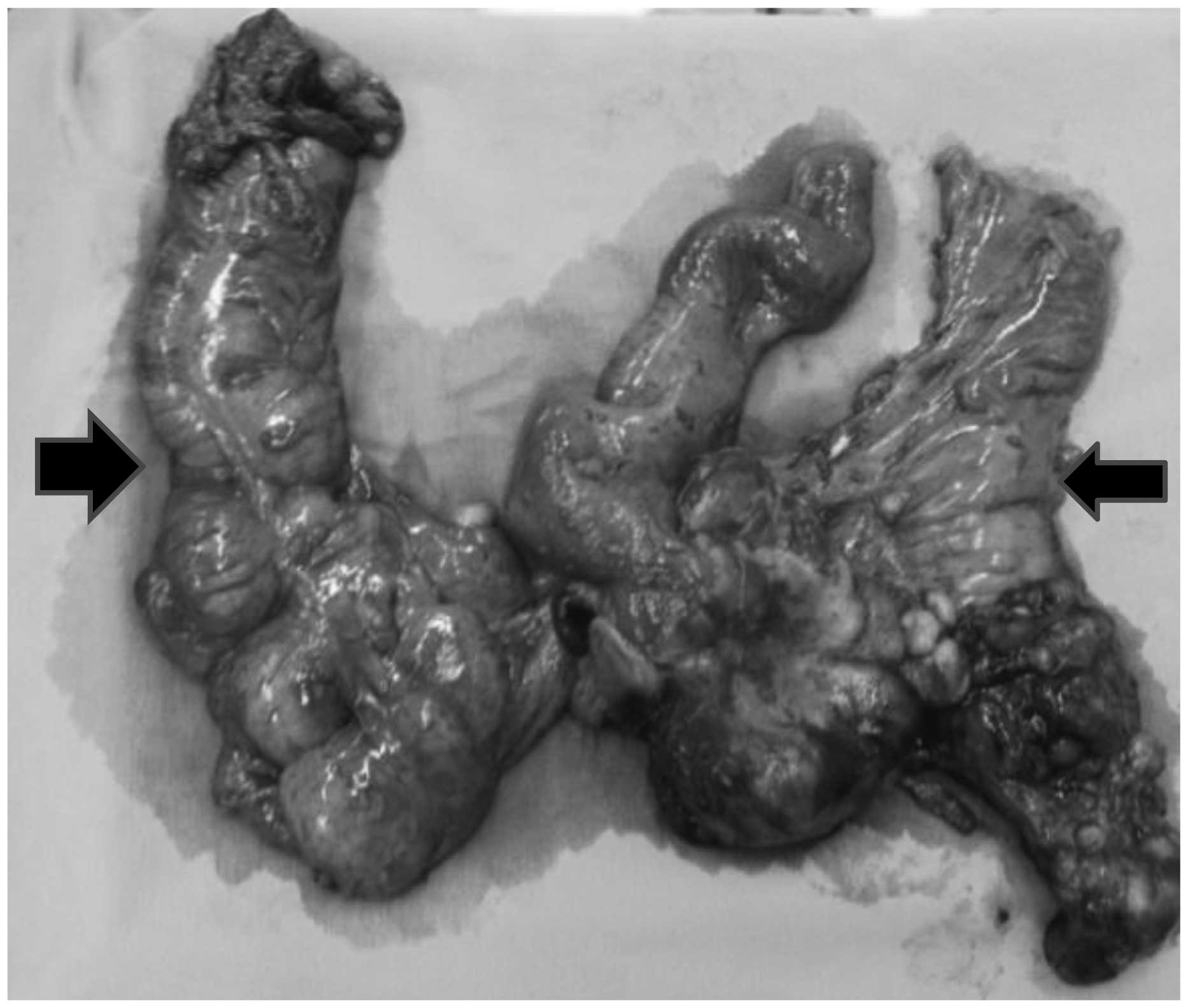
Analysis of the surgical specimen confirmed the
diagnosis of DSRCT. The morphological and immunohistochemical
findings were identical to those observed in the first biopsy.
Histological sections indicated a high-grade malignant neoplasm,
represented by small cells with hyperchromic nuclei in the center
of desmoplastic stroma (Fig. 5).
Furthermore, immunohistochemical analysis identified positivity for
cytokeratin and desmin, and negativity for S-100, myogenin, Wilms'
tumor suppressor gene 1 (WT1), cluster of differentiation 45 (CD45)
and chromogranin antibodies. The CD45 negativity excluded a
diagnosis of lymphoma.
The patient underwent adjuvant abdominal
radiotherapy (dose, 4.5 Gy; duration, 3 months; however, after one
year of follow-up, relapse of the disease was observed in the
abdominal cavity. The disease relapse was not treated and the
patient succumbed to the disease three months after relapse.
Case 2
A 12-year-old female was admitted to the Clinic
Hospital of Botucatu School of Medicine in April 2013 due to
abdominal pain, emesis and loss of appetite. A physical examination
revealed that the patient was in a good general condition, with a
body mass index of 33.9 kg/m2 and no palpable abdominal
tumors. Upon cross-sectional abdominal CT scan, a soft-tissue mass
measuring 6.5 cm in diameter was identified posterior to the
pancreatic tail and the stomach, with no anatomical line between
the stomach and the splenic vein.
In addition, poorly delimited solid hepatic nodules
with peripheral contrast enhancement were identified. The largest
hepatic nodule measured 3.4 cm in diameter, and was located on
segment IV. Chest CT and bone scintigraphy were normal, and an
analysis of tumor markers detected 751 U/l lactate dehydrogenase
(normal range, 313–618 U/l), <1.2 mU/ml β-human chorionic
gonadotropin (normal range, <5.00 mU/ml), 1.24 ng/ml
carcinoembryonic antigen and 2.89 ng/ml α-fetoprotein. During video
laparoscopy, a large pancreatic mass, multiple liver metastases and
ascites were identified. Biopsies were performed on the pancreatic
mass and liver metastases, and ascites fluid was collected.
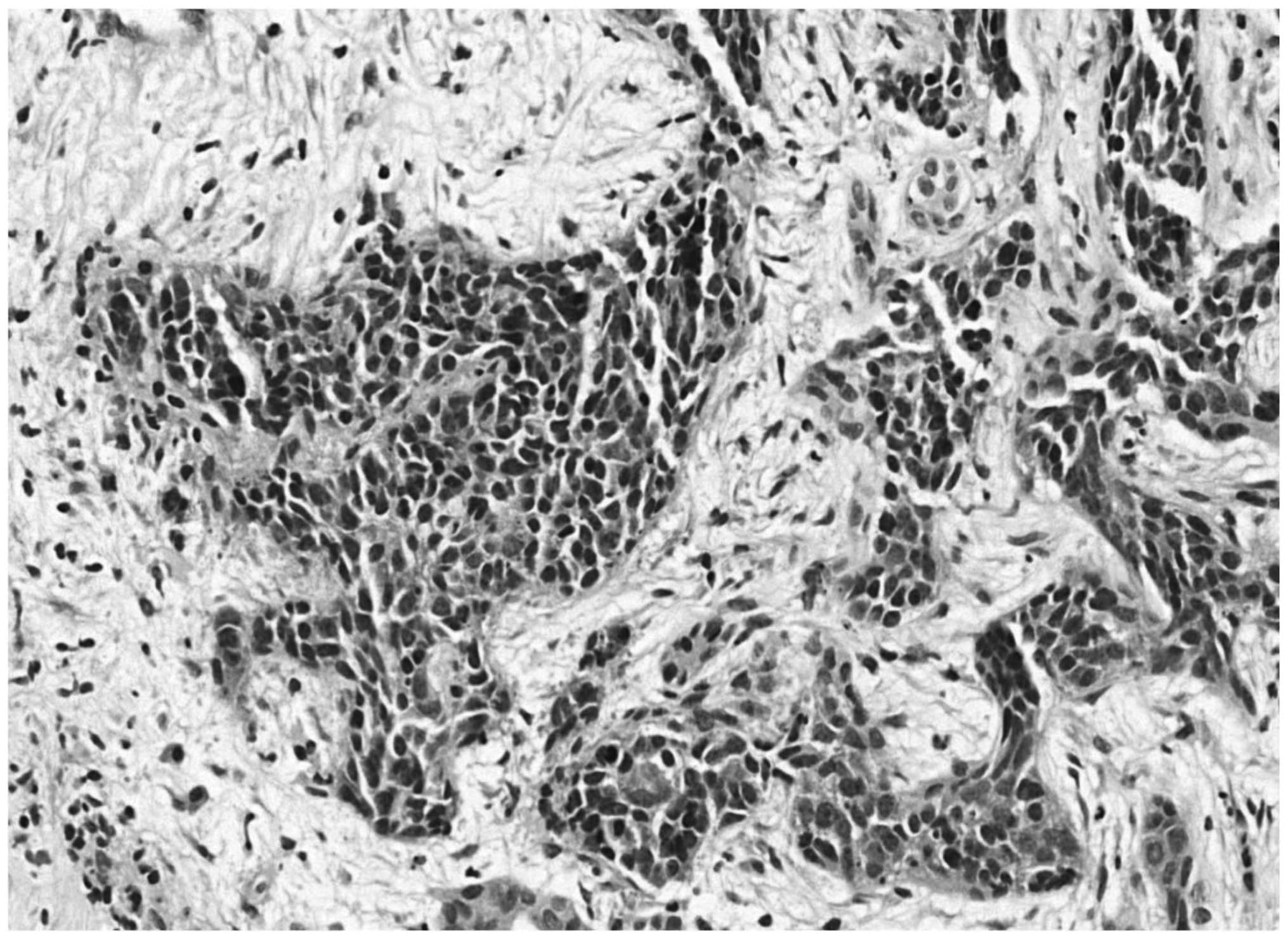
Subsequent histopathological analysis determined a malignant
neoplasm composed of small, blue, round cells, and
immunohistochemistry identified cytokeratin and vimentin expression
(with reinforcement in the paranuclear-Golgi zone), in addition to
positive focal staining of desmin in a typical dot-like pattern
(Fig. 6). Thus, the diagnosis of a
small cell tumor was determined. Additionally, the ascites fluid
was positive for neoplastic cells.
Due to extensive disease, chemotherapy was
scheduled. The treatment consisted of a vincristine, Adriamycin®
and cyclophosphamide (VAC) chemotherapeutic protocol (1
mg/m2 vincristine, 60 mg/m2 Adriamycin and
1.5 g/m2 cyclophosphamide) administered as intravenous
bolus infusion on day 1; after 21 days, an ifosfamide, carboplatin
and etoposide protocol (3.0 g/m2/day ifosfamide, 450
mg/m2 carboplatin and 150 mg/m2 etoposide)
was administered during 3 days. The course was repeated every 3
weeks. Following six sessions of chemotherapy, 25 sessions of
radiation therapy were scheduled (180 Gy/session; total dose, 4,500
Gy). While undergoing radiotherapy, Adriamycin was replaced by
actinomycin (1.25 mg/m2). Thus far, the patient has
completed 8 sessions of chemotherapy and is currently asymptomatic
with no abdominal complaints.
An abdominal CT scan revealed a small area (1.0×1.0
cm) in the pancreatic tail with no intravenous contrast
enhancement, and two small oval masses located in abdominal
segments IV and II, measuring 0.6–0.8 cm in diameter. A total of 25
sessions of chemotherapy, which started in April 2013, were
scheduled for completion of the treatment.
Written informed consent was obtained from the two
patients for participation in the present study.
Discussion
A diagnosis of DSRCT should be considered in
adolescents or young adults who present abdominal distention or an
abdominal mass, with abdominal or back pain, signs of
gastrointestinal obstruction, lack of appetite, ascites, anemia
and/or cachexia. The tumor is most commonly located in the
peritoneal cavity. Furthermore, DSRCT must be histologically and
cytologically distinguished from other small round cell tumors in
children and adolescents. For example, DSRCT should be
differentiated from rhabdomyosarcoma, non-Hodgkin's lymphoma,
Ewing's sarcoma, primitive neuroectodermal tumor, Wilms' tumor,
neuroblastoma and malignant mesothelioma (3,6).
Clinical findings associated with DSRCT include
ascites and intraparenchymal liver metastases, and less commonly,
retroperitoneal lymphadenopathy, hydronephrosis, bowel
calcifications and peritoneal nodular thickening (8). Non-specific symptoms are also observed,
such as pain, abdominal distension, and palpable abdominal, pelvic
or scrotal masses, occasionally associated with ascites (8).
Patients with DSRCT typically present with a short
duration of these symptoms and the disease is almost uniformly
fatal, regardless of the treatment modality administered. DSRCTs
are chemosensitive tumors, however, systemic chemotherapy typically
results in a short-lasting response and a poor gain in survival
time (9). Due to its refractory
response to individual treatment modalities and the aggressive
nature of the disease, an accurate diagnosis of DSRCT has
therapeutic implications and is therefore the primary goal of
clinicians (6,9).
The most characteristic feature of DSRCT in
cross-sectional imaging is single or multiple peritoneal
soft-tissue masses with no apparent organ of origin. Such imaging
may provide useful data regarding the tumor site and size, and the
efficacy of treatment. Imaging examination techniques for DSRCT
include ultrasound, CT, magnetic resonance imaging and
fluorodeoxyglucose-positron emission tomography/CT imaging. In
selected cases, immunohistochemical, electron microscopic,
molecular and genetic studies allow reliable discrimination of
these small cell neoplasms (10).
DSRCT is characterized by the following distinctive
pathological findings: A nesting pattern of cellular growth within
dense desmoplastic stroma, and immunohistochemical co-expression of
epithelial, muscle and neural markers (11). Of the 48 cases described by Zhang
et al (11), the tumor cells
exhibited diffuse to focal positivity for cytokeratin (37/42 cases;
88.10%), epithelial membrane antigen (33/41 cases; 80.49%), desmin
(45/46 cases; 97.83%), vimentin (43/45 cases; 95.56%), CD99 (6/20
cases; 30.00%), neuron-specific enolase (38/45 cases; 84.44%),
synaptophysin (2/15 cases; 13.33%) and chromogranin antibody (4/19
cases; 21.05%). The stromal cells of the tumor were positive for
smooth muscle antibody (10/13 cases; 76.92%) and HBME1 (2/2 cases;
100.00%). Therefore, DSRCT has a divergent differentiation, which
is an important feature of this tumor. Chang (12), in a review of the literature,
demonstrated that the tumor cells are positive for epithelial
(keratin and epithelial membrane antigen), mesenchymal (vimentin),
myogenic (desmin) and neural (neuron-specific enolase and CD56)
antibodies. The author also indicated that the majority of DSRCTs
are positive for WT-1, when the polyclonal antibody against the
amino terminus of the WT-1 protein is used. Furthermore, CD99
usually demonstrated cytoplasmic staining, as opposed to the
membranous staining observed in Ewing sarcoma/peripheral
neuroectodermal tumor (12).
Constitutive genetic expression observed in DSRCT
reveals the unique t(11;22)(p13;q11 or q12) reciprocal
translocation, the result of fusion between exon 7 of the Ewing's
sarcoma gene (EWS) on chromosome 22 with exon 8 of the WT1 gene on
chromosome 11. The EWS-WT1 fusion protein gene serves as a
disease-specific marker, and as its defining cytogenetic
abnormality, yields a definitive diagnosis of DSRCT (13). Molecular evidence of t(11;22)(p13;q12)
was also demonstrated by Zhang et al (11) in a small proportion of the patient
cohort.
The efficacy of treatment strategies and the
prognosis of patients with DSRCT remains controversial, with no
standard management protocols established. This is, in part, due to
the clinically aggressive nature of the neoplasm. For example,
complete excision is often difficult to obtain due to the presence
of multiple implants in the peritoneum. The lack of established
standard treatment protocol is also associated with the limited
number of patients in all previously reported series. However, the
current literature indicates that an aggressive approach involving
total macroscopic excision of the tumors combined with radiation
and chemotherapy may provide the greatest opportunity for disease
control and disease-free survival (6,9,13,14). Thus,
the elimination of sarcoma tumors and metastases using physical
approaches is essential for durable responses (15).
Therapeutic DSRCT management remains a challenge,
with low efficacy responses despite the combination of aggressive
treatment strategies, such as surgery, debulking, polychemotherapy,
whole abdominal radiation, hyperthermic intraperitoneal
chemotherapy (HIPEC), bone marrow transplantation and targeted
therapy (11,14).
In the retrospective study of 48 patients by Zhang
et al (11), the percentage of
patients who received surgery, complete resection or chemotherapy
was 79.17, 37.50 and 52.08%, respectively. The median follow-up
duration was 2.67 years, the median overall survival time was 24.33
months [95% confidence interval (CI), 9.74–38.92] and the median
event-free survival time of all patients was 8.00 months (95% CI,
5.13–10.89). Univariate analysis of this data revealed that
surgery, effective debulking surgery, chemotherapy and any two or
more combined therapies were significant prognostic factors for a
longer overall survival time (P<0.05).
Aggressive surgical debulking is the primary
therapeutic strategy for patients with DSRCT. Debulking surgery is
defined as the definitive removal of ≥90% of the tumor burden, as
complete resection is rarely possible due to extensive
dissemination. In the study by Zhang et al (11), 68.75% (33/48 cases) of patients
succumbed between 2 and 123 months (mean survival, 13.63
months).
In case 1 of the present study, the right ureter was
involved with extensive hydronephrosis; therefore, the selected
treatment strategy was surgical debulking of the tumor. Debulking
was performed in conjunction with block resection of the lesion
associated with a pelvic peritoniectomy and followed by
post-operative radiotherapy. However, due to extensive disease in
the second patient, chemotherapy with a VAC protocol and radiation
therapy were scheduled. A relapse of the disease was observed in
the abdominal cavity of the patient after one year; however, the
patient from case 2 is currently asymptomatic.
The most representative chemotherapeutic protocol
for patients with DSRCT is the P6 regimen, initially reported in
1996 by Kushner et al (9).
According to a subsequent study conducted by Lal et al
(5), 44% of patients underwent
induction of P6 chemotherapy, surgical debulking and radiotherapy.
The 3- and 5-year overall survival rates were 44 and 15%,
respectively. In addition, the three-year survival rates were 55%
for those receiving chemotherapy, surgery and radiotherapy, versus
27% when all three modalities were not used (P<0.020).
A metastatic seeding pattern via lymphatic and
hematogenous routes is common in DSRCT, with the omentum frequently
affected, followed by spread to distant lymph nodes, the liver, the
lungs and occasionally, other locations. Such events mean all
necessary efforts should be made to administer combined treatment
approaches to patients with this disease (15).
The effect of a complete resection of disseminated
DSRCTs on survival remains unknown due to the rarity of achieving
it during surgery (15). Therefore,
multimodal treatment in the form of high-dose (P6 protocol)
chemotherapy, maintenance chemotherapy, debulking surgery,
cytoreductive surgery and radiotherapy are generally preferred, as
these strategies have previously exhibited a tumor response.
Alternative treatment strategies include hemopoietic stem cell
transplantation, intensity-modulated radiotherapy, radiofrequency
ablation and HIPEC (15).
Biswas et al (16) demonstrated that complete surgical
excision appears to improve survival in patients with DSRCT;
however, additional adjuvant therapy is urgently required due to
the high recurrence and aggressive biology of the tumor (17). Bisogno et al (18) proposed a sequential intensified
chemotherapeutic strategy with peripheral blood stem cell (PBSC)
rescue for children and adolescents with DSRCT. The study was
designed to investigate the role of early sequential intensified
RMS 4.99 chemotherapy with PBSC rescue in soft-tissue sarcoma
patients with a poor prognosis. However, the prognosis for
pediatric patients with DSRCT did not improve following
administration of intensified chemotherapy early in the treatment
regime; therefore, the development of novel strategies is
required.
A complete surgical resection with cisplatin-based
microspheres in yttrium and HIPEC for cases with liver metastases
was identified to be useful in the treatment of DSRCT (17). Furthermore, it has been stated that
HIPEC is safe for use in children and may prolong disease-free
survival in select cases (19). In
addition, preclinical studies have demonstrated that vascular
endothelial growth factor receptor-2 (VEGFR-2) and VEGFA are
overexpressed in DSRCT, and that DSRCT xenografts are highly
responsive to treatment with anti-VEGF agents, such as bevacizumab.
However, data regarding the potential therapeutic role of
antiangiogenic agents in DSRCT is rare (14). Another candidate for the treatment of
DSRCT is sunitinib, a multi-kinase inhibitor that blocks various
tyrosine kinase receptors, such as VEGFR, platelet-derived growth
factor receptors, v-kit Hardy-Zuckerman 4 feline sarcoma viral
oncogene homolog, fms-related tyrosine kinase 3 and colony
stimulating factor receptor-1. Preliminary results build on a prior
report of a single patient with DSCRT responding to sunitinib,
indicating the potential efficacy of this agent, even in heavily
pretreated patients (20).
In conclusion, based on the analysis of the two
cases described in the present study, it was determined that
aggressive treatment regimens may induce tumor regression. However,
relapse of the disease is frequent and long-term survival is rare
with the currently available therapies. Considering that all
knowledge of this disease entity is based on reports in a small
number of patients, additional studies addressing the current
protocols are required to enable selection of the most appropriate
treatment regimens.
References
|
1
|
Gerald WL and Rosai J: Case 2.
Desmoplastic small cell tumor with divergent differentiation.
Pediatr Pathol. 9:177–183. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gerald WL, Miller HK, Battifora H, et al:
Intra-abdominal desmoplastic small round-cell tumor. Report of 19
cases of a distinctive type of high-grade polyphenotypic malignancy
affecting young individuals. Am J Surg Pathol. 15:499–513. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dufresne Al, Cassier P, Couraud L, et al:
Desmoplastic small round cell tumor: Current management and recent
findings. Sarcoma. 2012:7149862012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Syed S, Haque AK, Hawkins HK, Sorensen PHB
and Cowan DF: Desmoplastic small round cell tumor of the lung. Arch
Pathol Lab Med. 126:1226–1228. 2002.PubMed/NCBI
|
|
5
|
Lal DR, Su WT, Wolden SL, et al: Results
of multimodal treatment for desmoplastic small round cell tumors. J
Pediatr Surg. 40:251–255. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stuart-Buttle CE, Smart CJ, Pritchard S,
Martin D and Welch IM: Desmoplastic small round cell tumour: A
review of literature and treatment options. Surg Oncol. 17:107–112.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fletcher CDM, Bridge JA, Hogendoorn PCW
and Martens F: WHO Classification of Tumors of Soft Tissue and Bone
- Pathology and GeneticsIARC; Lyon, France: 2013
|
|
8
|
Gil A, Gomez Portilla A, Brun EA and
Sugarbaker PH: Clinical perspective on desmoplastic small
round-cell tumor. Oncology. 67:231–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kushner BH, LaQuaglia MP, Wollner N,
Meyers PA, Lindsley KL, Ghavimi F, et al: Desmoplastic small
round-cell tumor: Prolonged progression free survival with
aggressive multimodality therapy. J Clin Oncol. 14:1526–1531.
1996.PubMed/NCBI
|
|
10
|
Meis-Kindblom JM, Stenman G and Kindblom
LG: Differential diagnosis of small round cell tumors. Semin Diagn
Pathol. 13:213–241. 1996.PubMed/NCBI
|
|
11
|
Zhang J, Xu H, Ren F, Yang Y, Chen B and
Zhang F: Analysis of clinicopathological features and prognostic
factors of desmoplastic small round cell tumor. Pathol Oncol Res.
20:161–168. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang F: Desmoplastic small round cell
tumors: cytologic, histologic, and immunohistochemical features.
Arch Pathol Lab Med. 130:728–732. 2006.PubMed/NCBI
|
|
13
|
Quaglia MP and Brennan MF: The clinical
approach to desmoplastic small round cell tumor. Surg Oncol.
9:77–81. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Anderson PM and Pearson M: Novel
therapeutic approaches in pediatric and young adult sarcomas. Curr
Oncol Rep. 8:310–315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Talarico F, Iusco D, Negri L and Belinelli
D: Combined resection and its multi-agent adjuvant chemotherapy in
intra-abdominal desmoplastic small round cell is tumour: Case
report and review of the literature. G Chir. 28:367–370.
2007.PubMed/NCBI
|
|
16
|
Biswas G, Laskar S, Banavali SD, Gujral S,
Kurkure AP, Muckaden M, Parikh PM and Nair CN: Desmoplastic small
round cell abdominal tumour: Extra abdominal and abdominal
presentations and the results of treatment. Indian J Cancer.
42:78–84. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hayes-Jordan A and Anderson PM: The
diagnosis and management of desmoplastic small round cell tumor: A
review. Curr Opin Oncol. 23:385–389. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bisogno G, Ferrari A, Rosolen A, Alaggio
R, Scarzello G, Garaventa A, Arcamone G and Carli M: Sequential
intensified chemotherapy with stem cell rescue for children and
adolescents with desmoplastic small round-cell tumor. Bone Marrow
Transplant. 45:907–911. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hayes-Jordan A, Green H, Fitzgerald N,
Xiao L and Anderson P: Novel treatment for desmoplastic small round
cell tumor: Hyperthermic intraperitoneal perfusion. J Pediatr Surg.
45:1000–1006. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Italiano A, Kind M, Cioffi A, Maki RG and
Bui B: Clinical activity of sunitinib in patients with advanced
desmoplastic round cell tumor: A case series. Target Oncol.
8:211–213. 2013. View Article : Google Scholar : PubMed/NCBI
|