Introduction
Acute promyelocytic leukemia (APL) is a distinctive
subtype of acute myeloid leukemia that exhibits specific
cytogenetic features characterized by the reciprocal translocation
between chromosomes 15 and 17, which leads to the formation of the
PML-RARα fusion gene. With the encoded gene product, this fusion
gene interferes with the maturation process of myeloid cells and
finally results in phenotypic alterations in the target cells.
Prior to receiving all-trans retinoic acid (ATRA)-based regimens,
the complete remission rate for APL is 75–80% and the five year
disease-free survival rate is 35–45% (1). Despite a high complete remission rate of
95–95% and significant improvement in the five-year disease free
survival rate (to 74%) of patients with APL following treatment
with ATRA and arsenic trioxide (ATO) (1), certain patients inevitably relapse, even
subsequent to consolidation or maintenance therapies. Similar to
other variants of myeloid leukemia, the most frequent location of
relapse for APL is the bone marrow (BM); however, an increasing
frequency of extramedullary (EM) recurrence, mainly in the CNS and
skin, has been reported (2). Factors
associated with CNS relapse include advanced patient age, B-cell
antigen receptor isoform (3),
development of differentiation syndrome, presence of leukocytosis
at presentation and occurrence of hemorrhage in the CNS during
induction therapy (3). The present
study reports the case of a patient with APL.
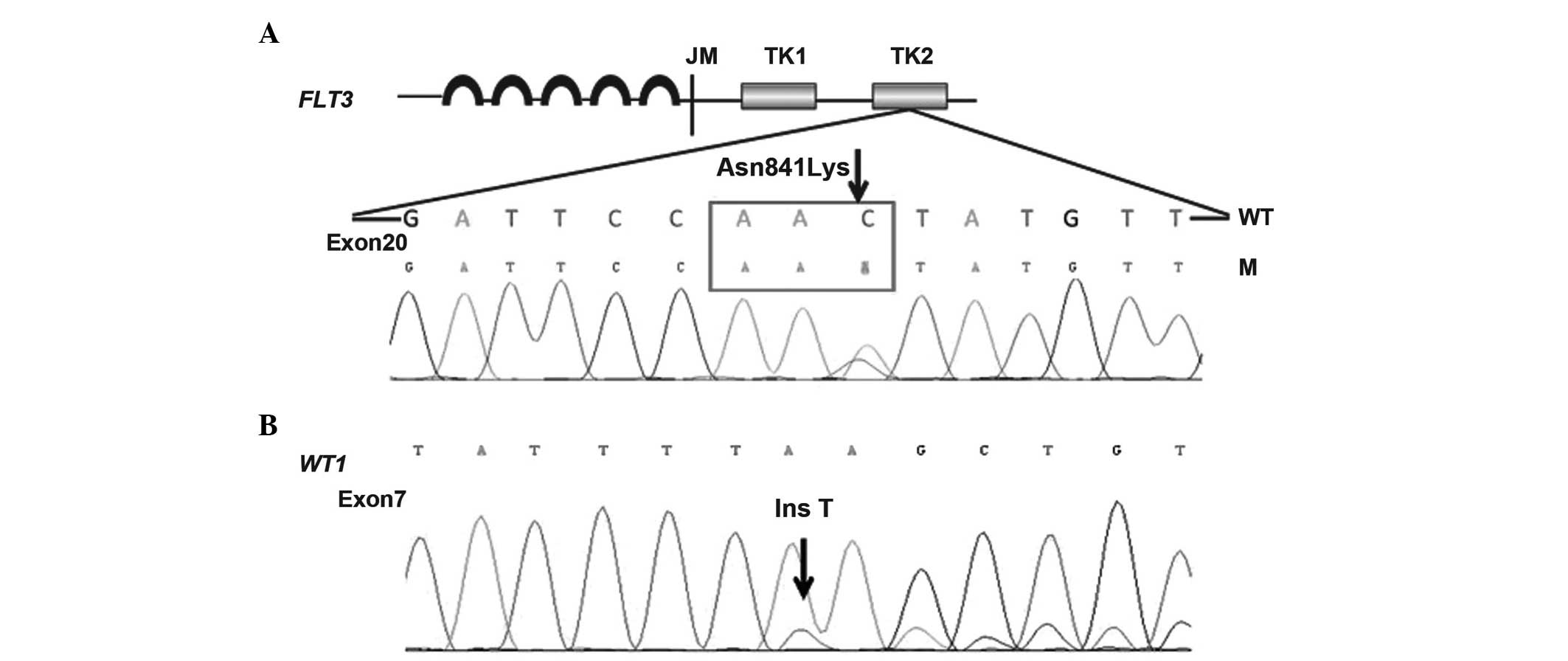
The patient exhibited a rare point mutation
(p.Asn841Gly, c.2523C>A) in the tyrosine kinase domain (TKD) of
the FMS-like tyrosine kinase 3 (FLT3-TKD) gene and a novel
Wilm tumor (WT1) gene mutation (c.1209_1210insT/p.K404X).
The patient suffered two CNS and systemic relapses during systemic
and intrathecal chemotherapy.
Case report
A 34-year-old Chinese man presented to the
Department of Hematology of West China Hospital, Sichuan University
(Chengdu, China) with a fever and chills in July 2011. The initial
laboratory evaluations revealed a white blood cell (WBC) count of
55.61×109 cells/l (normal range, 4–10×109
cells/l), with 85% abnormal promyelocytes (normal proportion, 0%),
a hemoglobin (HB) concentration of 111 g/l (normal range, 120–160
g/l) and a platelet (PLT) count of 62×109 cells/l
(normal range, 100–300×109 cells/l). The prothrombin
time (PT; normal range, 9.6–12.8 sec) and activated partial
thromboplastin time (APTT; normal range, 20–40 sec) at the time of
admission were 17.2 sec and 23.6 sec, respectively. The PT
international normalized ratio (PT INR) was 1.61 (normal range,
0.86–1.14) and the thrombin time (TT) was 24.7 sec (normal range,
14–22 sec). The fibrinogen level (FIB) was 0.65 g/l (normal range,
2–4g/l) and the D-dimer value was 35.56 mg/l fibrinogen equivalent
units (FEU; normal range, <0.55mg/l FEU), which indicated the
presence of disseminated intravascular coagulation (DIC). The BM
aspirate containing hypergranular promyelocytes was consistent with
the diagnosis of APL. Immunophenotyping of the BM sample revealed
that leukemic cells were positive for the expression of cluster of
differentiation (CD)13, CD33 and CD117, but did not express CD34,
CD14, CD36, CD5, CD7, CD56 or CD19. Human leukocyte antigen-DR and
CD64 were expressed at a low level. The gene mutational analysis
revealed the presence of the PML-RARα fusion gene, which was the
result of a t (15;17) translocation.
The (ATRA; 20 mg twice a day) therapy was initiated
immediately subsequent to the suspected diagnosis of APL. The
presence of the PML-RARα fusion gene was then rapidly confirmed in
the BM sample. Daunorubicin (DNR; 60 mg/d for 3 consecutive days)
and ATO (10 mg/d for 22 days) were added to the induction therapy
regimen. With nearly 40 days of induction therapy, the BM aspirate
indicated morphological complete remission, but the PML-RARα fusion
gene remained detectable. Following an additional two cycles of the
standard darubicin and Ara-C (IDA) regimen (20 mg idarubicin on day
1 and10 mg idarubicin on days 2 and 3; 150 mg Ara-C on days 1–7)
and one cycle of the intermediate-dose Ara-C regimen (40 mg DNR on
days 1 and 2; 1,800 mg Ara-C on days 1–4) as consolidation therapy,
the patient achieved complete molecular remission. Subsequently,
regimens containing ATO (10 mg/day ATO for 14 consecutive days),
ATRA (10 mg ATRA three times a day for 28 days), 6-mercaptopurine
(6-MP; 50 mg 6-MP three times a day, for 28 days) and methotrexate
(MTX; 15 mg MTX weekly for 4 weeks) were administered in succession
as maintenance therapy every three months. The cerebrospinal fluid
(CSF) was also monitored and intrathecal prophylaxis of
dexamethasone, MTX or Ara-C was performed regularly. Although
allogenic hematopoietic stem cell transplantation (allo-HSCT) was
then considered as an appropriate approach to improve the outcome
of the patient, a fully matched unrelated donor was not available
and the patient was unwilling to undergo haplo-identical
transplantation.
Routine CSF monitoring 20 months subsequent to the
diagnosis of APL revealed an elevated nucleated cell count
(526×106 cells/l) and an increased CSF total protein
level (0.52 g/l), with an elevated CSF pressure of 240 mm
H2O. The atypical promyelocytes that expressed CD117 and
CD33, but did not express CD5, were detected by flow cytometric
analysis. The patient exhibited no symptoms of CNS involvement at
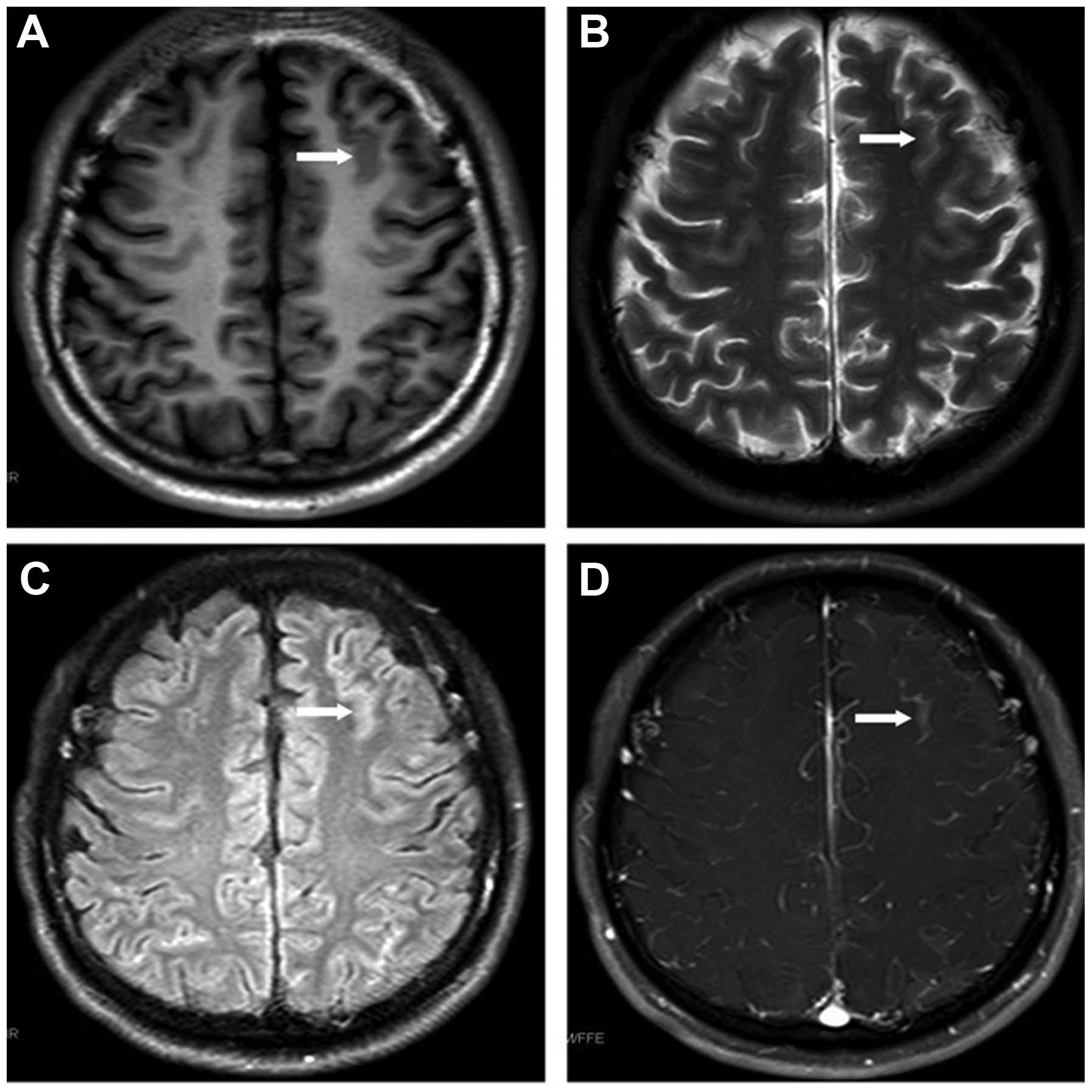
that time. Magnetic resonance imaging (MRI) revealed leukemic
linear infiltration at the frontal lobe (Fig. 1) and cerebellar tonsillar hernia. The
patient was therefore treated with 4 cycles of intrathecal
chemotherapy composed of 5 mg dexamethasone, 15 mg MTX and 50 mg
Ara-C, and 14 cycles of whole-brain and spinal cord radiotherapy,
followed by one cycle of the intermediate-dose Ara-C regimen (10 mg
idarubicin on days 1 and 2; 1,500 mg Ara-C on days 1–4). CSF
analysis eventually revealed a normal result, and the blood and BM
examinations also indicated complete hematological remission at
that time. Within three months of the CNS relapse and the
administration of the corresponding therapies, the bone marrow
aspirate indicated complete molecular remission, with the PML-RARα
fusion gene being undetectable. MRI revealed that the lesions
within the frontal lobe had diminished.
However, the patient developed systemic relapse with
persistent hyperleukocytosis and DIC only five months subsequent to
the first CNS relapse. Laboratory tests revealed a WBC count of
1.59×109 cells/l with 75% abnormal promyelocytes, APTT
of 68.7 sec, TT of 27.6 sec, FIB level of 0.73 g/l, and D-dimer
value of 29.73 mg/l FEU. The administration of 20 mg DNR daily for
6 consecutive days, 20 mg ATRA twice a day, and 10 mg ATO daily was
used as combined re-induction therapy, and the patient achieved
complete hematological remission again in almost one month,
demonstrating normal blood and BM tests and the PML-RARα fusion
gene was undetectable again. The patient subsequently received four
cycles of consolidation therapy, including the standard IDA regimen
and routine intrathecal chemotherapy.
Possible genetic changes were suspected due to the
CNS and systemic relapses observed in the present patient.
Therefore, 11 prognosis-associated genes, consisting of
CEBPA, DNAT3A, FLT3-ITD/TKD, IDH1,
IDH2, KIT, KRAS, NPM1, NRAS,
TET2 and WT1, were retrospectively analyzed. A rare
point mutation in the FLT3-TKD gene (p.Asn841Gly,
c.2523C>A) and a novel mutation of the WT1 gene
(c.1209_1210insT/p.K404X) were detected in the preserved bone
marrow and blood samples of the patient throughout the course of
disease (Fig. 2).
The patient suffered systemic relapse and DIC again
16 months after the intial CNS relapse. Laboratory tests revealed a
HB level of 126 g/l, PLT count of 36×109 PLT/l, WBC
count of 2.43×109 cells/l, PT of 18.5 sec, PT INR of
1.61, APTT of 41.9 sec, TT of 36.7 sec, FIB level of 0.50 g/l and
D-dimer value of 38 mg/l FEU. The BM smear revealed that 17.5% of
promyelocytic cells were abnormal, with a PML-RARα gene expression
rate of 16.39%. The patient received combined re-induction therapy
regimens consisting of 10 mg IDA on days 1–4, 20 mg ATRA twice a
day, and 10 mg ATO once a day for nearly 40 days. The patient
achieved hematological remission after this time period, with the
PML-RARα gene being expressed in 12.81% of BM cells and no symptoms
of CNS involvement being exhibited. However, routine CSF
examination revealed an elevated nucleated cell count of
185×106 cells/l, and 37.15% of cells were revealed to
possess the immunophenotype of abnormal promyelocytes, as
determined by flow cytometry. Considering the relapse and
refractory character of the present patient with APL and the
possible resistance of the APL to conventional therapy, alternative
induction and consolidation treatments composed of targeted FLT3
inhibitor, tetraarsenic tetrasulfide, intrathecal chemotherapy and
novel cytotherapy were administered in attempt to achieve an
improved therapeutic response. At present, the patient is being
prepared for ensuing salvage allo-HSCT.
Discussion
The long-term survival of the majority of patients
with APL improves subsequent to ATRA and ATO mediated treatment,
but certain patients inevitably relapse during the follow-up
period. The majority of post-remission relapses occur in the bone
marrow; however, ~3% present at EM sites, mainly the CNS and skin
(2). CNS relapse associated factors
include advanced age, specific BCR isoform (3), development of differentiation syndrome,
leukocytosis at presentation and hemorrhage in the CNS during
induction therapy (3). In previous
studies, treatment of APL with ATRA (4) and the occurrence of retinoic acid
syndrome (5) were considered to
predispose patients to EM relapse, but this was not reported by
other studies (6). Additionally, an
insufficient ATRA dose may fail to yield an effective concentration
in the EM sites, which is also a possible contributor to subsequent
relapse (7). With respect to
potential risk factors, certain studies have indicated that
FLT3-ITD mutations and an increased expression of adhesion
molecules, including CD56 (3), may
promote leukemic infiltration in the CNS. In the present study,
although the notable leukocytosis at presentation may be
responsible for the EM recurrence in the current patient, the
potential impact on the clinicobiological characteristics of APL
from two identified genetic mutations also requires
consideration.
FLT3, also termed CD135, is a member of the class
III tyrosine kinase receptor family, which also includes c-FMS,
c-KIT and PDGFR. FLT3 plays an important role in hematopoietic
malignancies and the FLT3 gene encodes a 993-amino acid
protein in humans, which comprises an immunoglobulin-like
extracellular ligand-binding domain, a transmembrane domain, a
juxtamembrane dimerization domain and a cytoplasmic domain with a
split tyrosine kinase motif. Two major forms of gain-of-function
mutations of FLT3, internal tandem duplication (ITD) and
point mutations within the activation loop of the tyrosine kinase
domain (TKD), were found in 21–38% and 9–20% of APL cases,
respectively (8–13). Among these mutations, Asp835Tyr
(D835Y) is the most frequent FLT3-TKD mutation [13.2% in
monocytic variant (M5); 11.8% in micro granular variant (M3v)] and
results in constitutive tyrosine phosphorylation, which boosts
neoplastic proliferation (14). In
contrast to common mutations, the rare mutation Asn841Gly (A841G),
which is located in the activation loop of FLT3-TKD2, was detected
in the present study. The activation loop usually forms a hydrogen
bond network together with adjacent amino acid residues; however,
the impact of Asn841Gly on the conformational change of the
activation loop, which leads to phosphatase activity variation in
FLT3-TKD has not been elucidated. In addition, a novel WT1
mutation (c.1209_1210insT/p.K404X), was also detected in the
present patient, but little is known about the prevalence of
WT1 mutations and their significance in APL; limited
information was found in a previous study, in which 4 out of 103
APL patients carrying WT1 mutations were predisposed to
leukemia relapse (15). Although
WT1 mutations act as well-recognized indicators of poor
prognosis, independent of chromosomal aberrations in acute myeloid
leukemia, this is not the same in FLT3-TKD, and their
prognostic significance in APL remains controversial (10,13,16–18).
The majority of the studies revealed that certain mutations
indicated inferior traits of APL. Since there is little information
regarding the association between genetic mutations and
extramedullary disease (EMD) in patients with APL (19), the possibility that the rare genetic
mutations observed in the present patient may be responsible for
the multiple CNS and systemic relapses could not be excluded.
At present, there has been no concensus on
prophylaxis for CNS relapse in APL (3,20). It is
recommended that intrathecal chemotherapy should be performed
subsequent to the first complete remission in patients at high CNS
relapse risk, including those with an elevated WBC count, low
platelet count, promyelocytes expressing CD56, differentiation
syndrome and high serum lactate dehydrogenase (3,21). The
polymerase chain reaction and fluorescence in situ
hybridization analyses of the CSF should also be performed to
detect any subclinical CNS involvement, which may indicate a
requirement for intrathecal chemotherapy (3). The present patient was administered with
regular intrathecal prophylaxis of dexamethasone, MTX and Ara-C and
routine monitoring of CSF examination. CNS relapse was confirmed by
flow cytometry of the CSF without presenting any clinical
manifestations each time.
Despite the impressive complete remission and
survival results of APL that have been achieved, the prognosis for
CNS relapse of APL patients is generally poor (1). In a previous study, 20/31 patients
relapsed. The duration of remission ranged between 1 and 144
months, with a median of 5 months (22). The present study was reported 20
months subsequent to the initial diagnosis. As it is challenging to
avoid CNS relapse and subsequent systemic relapse in patients with
APL that possess molecular abnormalities and leucocytosis,
regardless of routine prophylaxis for CNS involvement, the
remaining approach for APL patients with high CNS relapse risks may
be allo-HSCT immediately subsequent to achieving the first
hematological remission.
In summary, the present study reported the case of a
patient with APL that experienced multiple CNS relapses and
systemic relapses, and harbored unfavorable gene mutations. The
present study highlights the importance of carefully monitoring EMD
during follow-up, which may be a clinical biomarker of impending
systemic relapses, and the importance of risk stratification with
mutational analysis for APL. However, additional preclinical
studies investigating these novel mutations and the interpretation
of the mutation status of the genes during APL progression continue
to be required. In addition, it appears inevitable that patients
with APL that possess leucocytosis and unfavorable gene mutations
relapse even following treatment with ARTA, ATO and
anthracycline-containing systemic therapy in combination with
intrathecal prophylaxis, which indicates that the patients with APL
that possess a high relapse risk should receive immediate allo-HSCT
whenever possible.
Acknowledgements
The authors thank Dr Chun-rong Tong from the
Department of Immunotherapy and Wen Teng from the Department of
Laboratory Medicine (Lu Daopei Hematology and Oncology Center,
Beijing, China) for their helpful discussions and technical
assistance.
References
|
1
|
Wang ZY and Chen Z: Acute promyelocytic
leukemia: From highly fatal to highly curable. Blood.
111:2505–2515. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de Botton S, Sanz MA, Chevret S, Dombret
H, Martin G, Thomas X, Mediavilla JD, Recher C, et al European APL
Group; PETHEMA Group: Extramedullary relapse in acute promyelocytic
leukemia treated with all-trans retinoic acid and chemotherapy.
Leukemia. 20:35–41. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Colovic N, Tomin D, Vidovic A, et al:
Central nervous system relapse in CD56+,
FLT3/ITD+ promyelocytic leukemia. Med Oncol. 29:260–262.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wiernik PH, De Bellis R, Muxi P and
Dutcher JP: Extramedullary acute promyelocytic leukemia. Cancer.
78:2510–2514. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ko BS, Tang JL, Chen YC, et al:
Extramedullary relapse after all-trans retinoic acid treatment in
acute promyelocytic leukemia - the occurrence of retinoic acid
syndrome is a risk factor. Leukemia. 13:1406–1408. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Specchia G, Lo Coco F, Vignetti M, et al:
Extramedullary involvement at relapse in acute promyelocytic
leukemia patients treated or not with all-trans retinoic acid: A
report by the Gruppo Italiano Malattie Ematologiche dell'Adulto. J
Clin Oncol. 19:4023–4028. 2001.PubMed/NCBI
|
|
7
|
Ohno R, Asou N and Ohnishi K: Treatment of
acute promyelocytic leukemia: Strategy toward further increase of
cure rate. Leukemia. 17:1454–1463. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Callens C, Chevret S, Cayuela JM, et al
European APL Group: Prognostic implication of FLT3 and Ras gene
mutations in patients with acute promyelocytic leukemia (APL): A
retrospective study from the European APL Group. Leukemia.
19:1153–1160. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuchenbauer F, Schoch C, Kern W, et al:
Impact of FLT3 mutations and promyelocytic leukemia-breakpoint on
clinical characteristics and prognosis in acute promyelocytic
leukemia. Br J Haematol. 130:196–202. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barragán E, Montesinos P, Camos M, et al
PETHEMA Group; HOVON Group: Prognostic value of FLT3-mutations in
patients with acute promyelocytic leukemia treated with all-trans
retinoic acid and anthracycline monochemotherapy. Haematologica.
96:1470–1477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chillón MC, Santamaría C, García-Sanz R,
Balanzategui A, Sarasquete ME, Alcoceba M, Marín L, Caballero MD,
Vidriales MB, Ramos F, Bernal T, et al: Long FLT3 internal tandem
duplications and reduced PML-RARα expression at diagnosis
characterize a high-risk subgroup of acute promyelocytic leukemia
patients. Haematolgica. 95:745–751. 2010. View Article : Google Scholar
|
|
12
|
Gale RE, Hills R, Pizzey AR, Kottaridis
PD, Swirsky D, Gilkes AF, Nugent E, Mills KI, Wheatley K, Solomon
E, et al NCRI Adult Leukaemia Working Party: Relationship between
FLT3 mutation status, biologic characteristics and response to
targeted therapy in acute promyelocytic leukemia. Blood.
106:3768–3776. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schnittger S, Bacher U, Haferlach C, Kern
W, Alpermann T and Haferlach T: Clinical impact of FTL3 mutation
load in acute promyelocytic leukemia with t(15;17)/PML-RARA.
Haematologica. 96:1799–1807. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bacher U, Haferlach C, Kern W, Haferlach T
and Schnittger S: Prognostic relevance of FTL3-TKD mutations in
AML: The combination matters - an analysis of 3082 patients. Blood.
111:2527–2737. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gaur GC, Ramadan SM, Cicconi L, Noguera
NI, Luna I, Such E, et al: Analysis of mutational status, SNP
rs16754, and expression levels of Wilms tumor 1 (WT1) gene in acute
promyelocytic leukemia. Ann Hematol. 91:1855–1860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hou HA, Huang TC, Lin LI, Liu CY, Chen CY,
Chou WC, Tang JL, Tseng MH, Huang CF, Chiang YC, et al: WT1
mutation in 470 adult patients with acute myeloid leukemia:
Stability during disease evolution and implication of its
incorporation into a survival scoring system. Blood. 115:5222–5231.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shih LY, Kuo MC, Liang DC, Huang CF, Lin
TL, Wu JH, Wang PN, Dunn P and Lai CL: Internal tandem duplication
and Asp835 mutations of the FMS-like tyrosine kinase 3 (FLT3) gene
in acute promyelocytic leukemia. Cancer. 98:1206–1216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kutny MA, Moser BK, Laumann K, Feusner JH,
Gamis A, Gregory J, Larson RA, Powell BL, Stock W, Willman CL, et
al: FLT3 mutation status is a predictor of early death in pediatric
acute promyelocytic leukemia: A report from the Children's Oncology
Group. Pediatr Blood Cancer. 59:662–667. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tashiro H, Shirasaki R, Oka Y, Sugao T,
Mizutani-Noguchi M, Yamamoto T, Akiyama N, Kawasugi K and Shirafuji
N: FLT3 internal tandem duplication is associated with a high
relapse rate and central nervous system involvement in acute
promyelocytic leukemia cases: Single institutional analysis. Eur J
Haematol. 86:272–273. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Montesinos P, Díaz-Mediavilla J, Debén G,
Prates V, Tormo M, Rubio V, Pérez I, Fernández I, Viguria M, Rayón
C, et al: Central nervous system involvement at first relapse in
patients with acute promyelocytic leukemia treated with all-trans
retinoic acid and anthracycline monochemotherapy without
intrathecal prophylaxis. Haematologica. 94:1242–1249. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nagai S, Nannya Y, Arai S, Yoshiki Y,
Takahashi T and Kurokawa M: Molecular or cytogenetic monitoring and
preemptive therapy for central nervous system relapse of acute
promyelocytic leukemia. Haematologica. 95:169–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bae SH, Ryoo HM, Cho HS, Lee JL, Lee KH
and Hyun MS: Meningeal relapse in a patient with acute
promyelocytic leukemia: A case report and review of the literature.
J Korean Med Sci. 19:311–314. 2004. View Article : Google Scholar : PubMed/NCBI
|