Introduction
Chordomas are rare tumors that are believed to be
derived from the remnants of the embryonic notochord. They are
usually apparent along the axial side of the body, predominantly in
the skull base and sacral regions (1). The mean age at diagnosis is between 40
and 60 years, with a marginally younger mean age for cases at the
skull base (2). The tumor often
presents with cranial nerve dysfunction, and slow-growing but
infiltrative characteristics. In addition, the surrounding bones
and neurovascular structures are involved (3). The current approach of radical surgery
plus adjuvant radiotherapy has improved the outcome, however, the
majority of patients develop tumor recurrence and treatment
complications, resulting in a median survival period of 6.29 years,
with 5-, 10- and 20-year rates of 67.6, 39.9 and 13.1%,
respectively (2,4,5).
The incidence of the tumor is ~0.08/100,000
individuals (1–5) and little is known about its etiology. As
the majority of cases reported in the literature are sporadic,
familial chordoma cases with >3 patients identified within 1
family, involving >2 generations have been reported only 8 times
in the literature (Table I) (6–13). The
present study reports the case of a patient with familial skull
base chordoma and the details of 3 other pathologically confirmed
cases within the family, with a review of the literature.
 | Table I.Familial chordomas reported in the
literature. |
Table I.
Familial chordomas reported in the
literature.
| ID | Date (ref) | Number of
patients | Locations | Male:female | Age range, years | Mean age, years |
|---|
| 1 | 1958 (13) | 2 | Sacral | 1:1 | 52 | 52 |
| 2 | 1964a (12) | 2 | Nasopharynx | / | / | / |
| 3 | 1975 (9) | 3 | Nasopharynx,
clivus | 1:2 | 3–51 | 25 |
| 4 | 1998 (11) | 4 | Skull base,
sacral | 2:2 | 20–44 | 32 |
| 5 | 1999 (7) | 2 | Clivus | 1:1 | 8–12 | 10 |
| 6 | 2001 (8) | 10 | Skull base,
sacral | / | / | / |
| 7b | 2006 (18) | 8 | Skull base | 1:3 | / | / |
| 8b | 2006 (18) | 3 | Clivus | 1:2 | 3–55 | 30 |
| 9 | Present case | 4 | Clivus,
nasopharynx | 2:2 | 14–47 | 31 |
Case report
History and physical examination
In November 2011, a 15-year-old female with no other
medical history presented to the Department of Neurosurgery at
Beijing Tiantan Hospital (Beijing, China) with snoring and apnea
that had persisted for ~4 years, along with a headache that had
been present for ~6 months, which had been aggravated by symptoms
of nausea and vomiting for ~1 month. The patient had no history of
surgery or communicable diseases. On clinical examination, the
patient exhibited hoarseness. Eye movement was normal and there
were no signs of facial paralysis or dysaudia. No symptoms of
deglutition dysfunction were present. The patient exhibited good
strength in the limbs and the results of Romberg testing were
negative. The magnetic resonance imaging (MRI) and computed
tomography scans showed a bony invasive mass of the skull base
area, involving the nasal regions and clivus. The scans further
revealed that the lesion was compressing the brain stem from the
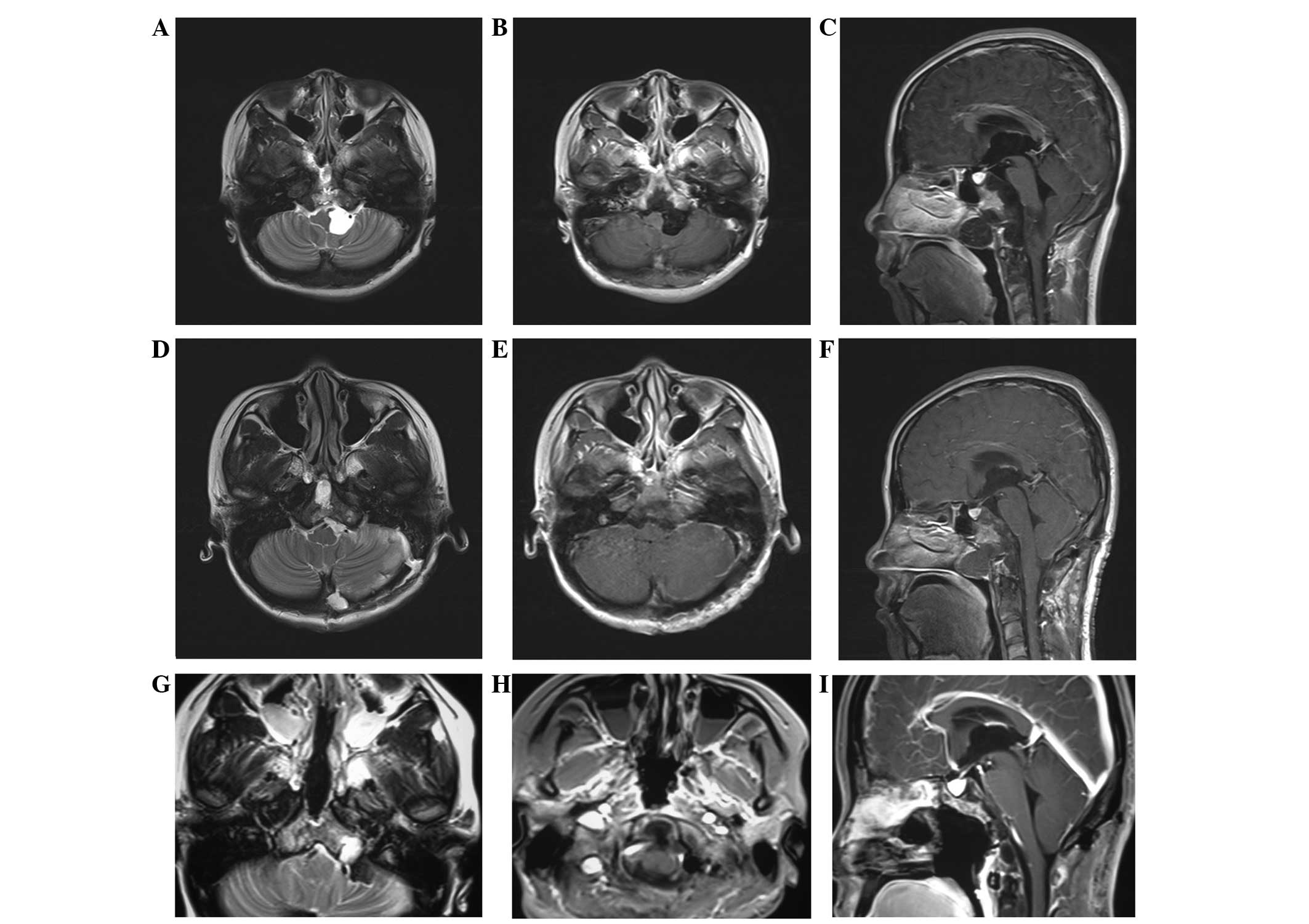
left side (Fig. 1A–C).
Surgery and post-operative course
A staged surgery was suggested, and a far lateral
approach was performed with a gross resection of the intracranial
mass, under electrophysiological surveillance. The post-operative
period was uneventful, with the exception of aggravation of the
hoarseness and sixth nerve palsy. The symptoms improved 6 months
later and MRI showed a good resection of the intracranial lesions
(Fig. 1D–F). The patient then
received the second surgery for the mass of the nasal region using
a subtotal resection (Fig. 1G–I).
Following the staged surgery, the patient exhibited no symptoms of
cerebrospinal fluid leakage, and the main symptoms of snoring and
apnea had almost disappeared. The patient received radiation
therapy (γ-knife, 28 Gy, one time) 1 year after the first surgery,
prior to returning back to school with no marked symptoms. At the
4-year follow-up, the patient exhibited no signs of recurrence, as
confirmed by MRI.
Pathological findings
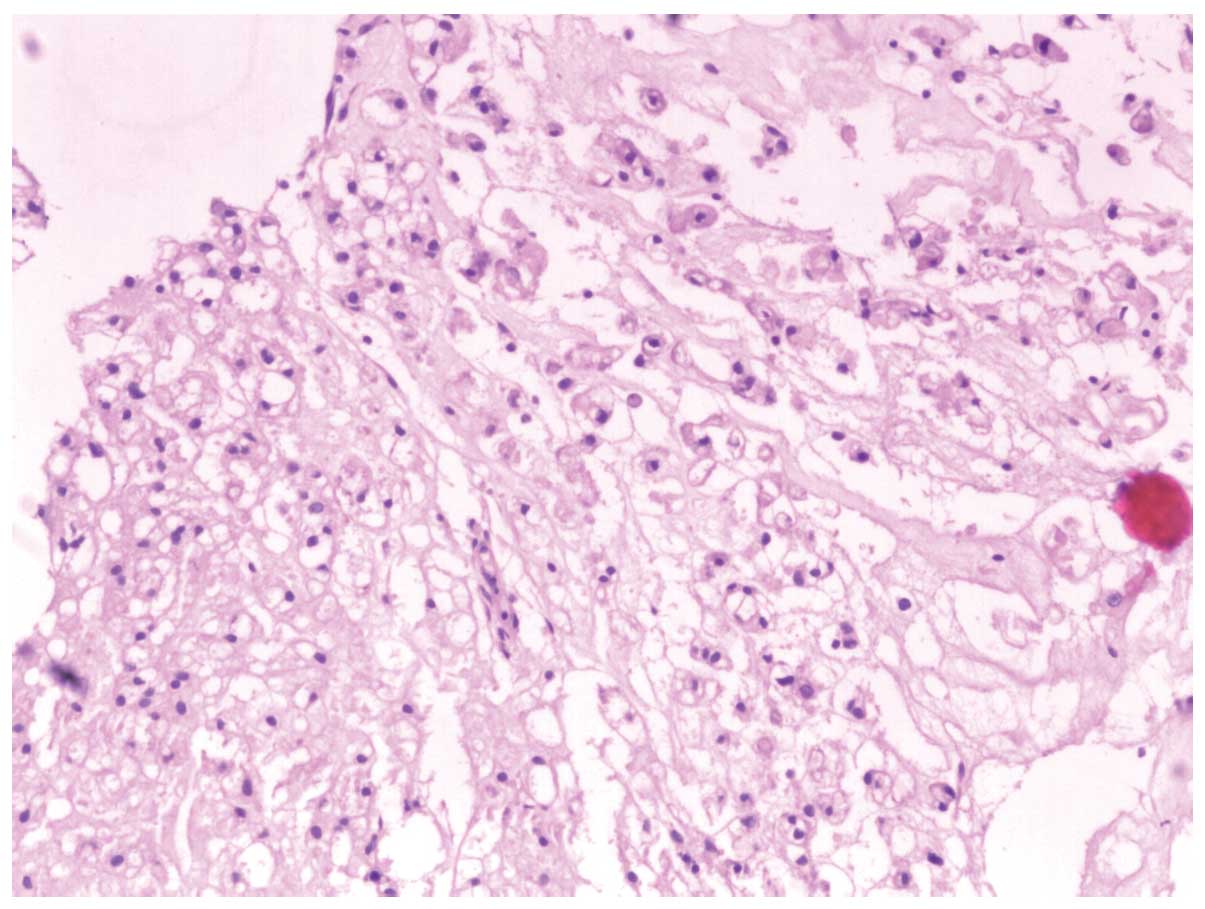
The tissue appeared reddish in color upon gross
inspection. Under the light microscope, the tumor was composed of
cords and strands of intermediate-sized tumor cells, showing
typical bubble-like cells with a bolus distribution. The
bubble-like cells were intermediate in size and round in shape,
with abundant cytoplasm. The nuclei were surrounded by cytoplasm
containing vacuoles (Fig. 2).
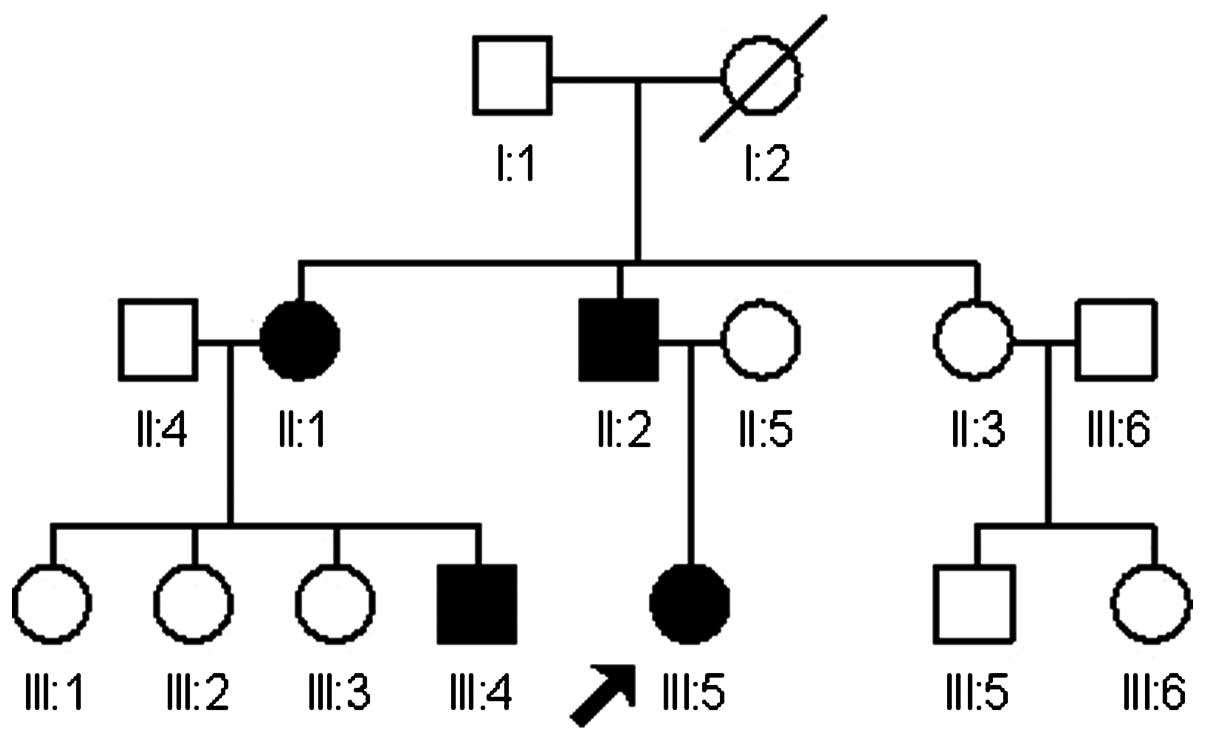
Family history
The patient had a strong familial history of
chordoma. At 6 months prior to presentation, the patient's father
(II:2) presented with a clivus chordoma at the age of 44 and
underwent removal via an endoscopic endonasal approach, followed by
radiation therapy. The patient's paternal aunt (II:1) had presented
with a clivus chordoma at the age of 47, which was also treated
with endoscopic endonasal therapy and confirmed to be a chordoma
pathologically. Furthermore, the son of this paternal aunt, an
18-year-old male (III:4) who also presented with symptoms of
snoring, underwent an epipharyngoscopy and was confirmed with
chordoma of the nasal region. Finally, the patient's paternal
grandmother (I:2) had succumbed from an unknown cause during the
fourth decade of life, with the symptoms of a chronic headache and
deglutition disability. Among the remaining paternal family
members, 3 members of a second aunt's family (II:3, III:5, III:6)
suffered from snoring, but did not receive any examination.
According to these clinical findings, a family pedigree was
achieved (Fig. 3).
Discussion
Familial chordomas are rare, and among the cases
discussed in the literature, only 8 familial chordomas have been
reported (Table I) (6–13). Between
1998 and 2011, ~256 patients with skull base chordomas have been
treated in the 7th Ward, Department of Neurosurgery, Beijing
Tiantan Hospital, Capital Medical University (Beijing, China)
(2–3).
Only one case presented with a familial history, so the estimated
incidence of familial chordomas was 0.4% (1/256) in all the
chordomas.
Chordoma is characterized as slow growing, often
with recurrence, mostly within 8 years (2–4). Studies
have indicated that a Hispanic ethnicity, a small tumor size, a
high socioeconomic status and surgical intervention are factors
that favor good survival, while adjuvant radiation therapy is a
controversial factor (14–16). In the present case, the diagnosis of a
chordoma was suspected at presentation and later confirmed
pathologically. When considering the radical resection of the
tumor, staged surgery plus adjuvant radiation (γ-knife) was
recommended for the 15-year-old female patient, and the outcome was
good at the time of follow-up. The results of the present case were
similar to a case reported by Chau et al (17), in which a combined endoscopic
endonasal and posterior cervical approach was used, together with
proton radiotherapy, in an 18-year-old male with a clivus chordoma
(17).
Reviewing the literature, a female predominance can
be found in the familial chordomas, with a male to female ratio of
1:1.8, which is the same as the gender difference in the skull base
chordomas (6–13,18,19).
Familial chordomas were most likely to occur in the area of the
skull base; among the 8 families identified, only two cases within
1 family were reported in the sacrococcygeal region in 1958
(19), while the other cases mainly
occurred in the skull base area (6–13),
including the present case. The familial chordomas may have
exhibited an early onset of symptoms, which made the age of
diagnosis much younger than that of the sporadic chordomas, with
the mean age of 29 and 40 years, respectively. More children and
adolescents were diagnosed in the familial chordomas (1,6–13,18,19). In
the present study, there were 2 adolescents (aged 15 and 18 years),
and 2 adults (aged 44 and 47 years), with a mean age of 31 years.
The younger generation of the family were diagnosed earlier than
the older generation, possibly due to earlier onset of symptoms as
well as better access to healthcare as a result of the improved
economy.
The majority of the studies concerning the genetic
mechanism of chordomas were primarily concerned with sporadic
chordomas. The cause of chordomas has been largely unknown,
however, gene deletions and gains have been noticed in the majority
of cases (20,21), and chromothripsis has become a great
focus of attention in the research of chordomas (22). The same is true in familial chordoma.
In 1998, Stepanek et al (11)
first suggested the autosomal dominant inheritance pattern.
Afterward, several studies using different methods analyzed the
gene abnormities in familial chordomas, and the 1p36.31–1p36.13 and
7q33 regions were found to be associated. However, the studies
failed to obtain a consensus (7,8,10,23). A
promising chordoma-specific gene, known as brachyury, localized in
6q27, was proved to be a key point in chordoma research during cell
line experiments (24–26). This gene was a member of T-box family,
containing a brachyury transcription factor, with a critical role
in notochord development (25). In
2009, Yang et al analyzed 8 familial chordomas using
high-resolution array-based comparative genomic hybridization and
combined genetic linkage analyses, suggesting that the T/brachyury
homolog was a major susceptibility gene for familial chordomas
(6). A recent study confirmed that an
allele at rs2305089 of the T gene, located in the exon area and
resulting in a Gly177Asp alteration, was strongly associated with
chordoma (27,28). However, the Gly177Asp single
nucleotide polymorphism site was not associated with chordomas in
the Han Chinese population studied (29).
In the current study, a family with 4 pathologically
confirmed skull base chordomas was presented. In general, familial
chordomas are predominantly located at the skull base and are
diagnosed at a younger age compared with sporadic chordoma. The
T/brachyury homolog gene may be a causative gene in familial and
sporadic chordomas, however, the genetic mechanism for chordomas
remains unclear. Further genetic studies and long term follow-up
are required for elucidation.
Acknowledgements
The authors would like to thank the patients for
their involvement in the present study and to all of those at
Beijing Tian Tan Hospital who contributed to the present study.
This study was supported in part by the Natural Science Foundation
of China (grant no. 81101910) and the Natural Science Foundation of
Beijing (grant no. 7142052).
References
|
1
|
McMaster ML, Goldstein AM, Bromley CM,
Ishibe N and Parry DM: Chordoma: Incidence and survival patterns in
the United States, 1973–1995. Cancer Causes Control. 12:1–11. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu Z, Zhang J, Zhang L, Jia G, Tang J,
Wang L and Wang Z: Prognostic factors for long-term outcome of
patients with surgical resection of skull base chordomas-106 cases
review in one institution. Neurosurg Rev. 33:451–456. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang L, Wu Z, Tian K, Li G and Zhang J:
Clinical and pathological features of intradural retroclival
chordoma. World Neurosurg. 82:791–798. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Di Maio S, Temkin N, Ramanathan D and
Sekhar LN: Current comprehensive management of cranial base
chordomas: 10-year meta-analysis of observational studies. J
Neurosurg. 115:1094–1105. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoch BL, Nielsen GP, Liebsch NJ and
Rosenberg AE: Base of skull chordomas in children and adolescents:
A clinicopathologic study of 73 cases. Am J Surg Pathol.
30:811–818. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang XR, Ng D, Alcorta DA, Liebsch NJ,
Sheridan E, Li S, Goldstein AM, Parry DM and Kelley MJ: T
(brachyury) gene duplication confers major susceptibility to
familial chordoma. Nat Genet. 41:1176–1178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dalprà L, Malgara R, Miozzo M, Riva P,
Volonte M, Larizza L and Conti AM Fuhrman: First cytogenetic study
of recurrent familial chordoma of the clivus. Int J Cancer.
81:24–30. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kelley MJ, Korczak JF, Sheridan E, Yang X,
Goldstein AM and Parry DM: Familial chordoma, a tumor of
notochordal remnants, is linked to chromosome 7q33. Am J Hum Genet.
69:454–460. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kerr WA, Allen KL, Haynes DR and Sellars
SL: Letter: Familial nasopharyngeal chordoma. S Afr Med J.
49:15841975.PubMed/NCBI
|
|
10
|
Miozzo M, Dalprà L, Riva P, Volontà M,
Macciardi F, Pericotti S, Tibiletti MG, Cerati M, Rohde K, Larizza
L and Conti AM Fuhrman: A tumor suppressor locus in familial and
sporadic chordoma maps to 1q36. Int J Cancer. 87:68–72. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stepanek J, Cataldo SA, Ebersold MJ,
Lindor NM, Jenkins RB, Unni K, Weinshenker BG and Rubenstein RL:
Familial chordoma with probable autosomal dominant inheritance. Am
J Med Genet. 75:335–336. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Enin IP: Chordoma of the nasopharynx in 2
members of a family. Vestn Otoinolaringol. 26:88–90. 1964.(In
Russian).
|
|
13
|
Foote RF, Ablin G and Hall WW: Chordoma in
siblings. Calif Med. 88:383–386. 1958.PubMed/NCBI
|
|
14
|
Lee J, Bhatia NN, Hoang BH, Ziogas A and
Zell JA: Analysis of prognostic factors for patients with chordoma
with use of the California cancer registry. J Bone Joint Surg Am.
94:356–363. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jian BJ, Bloch OG, Yang I, Han SJ, Aranda
D and Parsa AT: A comprehensive analysis of intracranial chordoma
and survival: A systematic review. Br J Neurosurg. 25:446–453.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu AL, Wang ZC, Sun SB, Wang MH, Luo B
and Liu P: Gama knife radiosurgery for residual skull base
chordomas. Neurol Res. 30:557–561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chau AM, Lazzaro A, Mobbs RJ and Teo C:
Combined endoscopic endonasal and posterior cervical approach to a
clival chordoma. J Clin Neurosci. 17:1463–1465. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bhadra AK and Casey AT: Familial chordoma:
A report of two cases. J Bone Joint Surg Br. 88:634–636. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bayrakli F, Guney I, Kilic T, Ozek M and
Pamir MN: New candidate chromosomal regions for chordoma
development. Surg Neurol. 68:425–430. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hallor KH, Staaf J, Jönsson G, Heidenblad
M, von Steyern F Vult, Bauer HC, Ijszenga M, Hogendoorn PC, Mandahl
N, Szuhai K and Mertens F: Frequent deletion of the CDKN2A locus in
chordoma: Analysis of chromosomal imbalances using array
comparative genomic hybridization. Br J Cancer. 98:434–442. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Le LP, Nielsen GP, Rosenberg AE, Thomas D,
Batten JM, Deshpande V, Schwab J, Duan Z, Xavier RJ, Hornicek FJ
and Iafrate AJ: Recurrent chromosomal copy number alterations in
sporadic chordomas. Plos One. 6:e188462011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stephens PJ, Greenman CD, Fu B, Yang F,
Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA,
et al: Massive genomic rearrangement acquired in a single
catastrophic event during cancer development. Cell. 144:27–40.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang X, Beerman M, Bergen AW, Parry DM,
Sheridan E, Liebsch NJ, Kelley MJ, Chanock S and Goldstein AM:
Corroboration of a familial chordoma locus on chromosome 7q and
evidence of genetic heterogeneity using single nucleotide
polymorphisms (SNPs). Int J Cancer. 116:487–491. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Presneau N, Shalaby A, Ye H, Pillay N,
Halai D, Idowu B, Tirabosco R, Whitwell D, Jacques TS, Kindblom LG,
et al: Role of the transcription factor T (brachyury) in the
pathogenesis of sporadic chordoma: A genetic and functional-based
study. J Pathol. 223:327–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jambhekar NA, Rekhi B, Thorat K, Dikshit
R, Agrawal M and Puri A: Revisiting chordoma with brachyury, a ‘New
Age’ marker: Analysis of a validation study on 51 cases. Arch
Pathol Lab Med. 134:1181–1187. 2010.PubMed/NCBI
|
|
26
|
Vujovic S, Henderson S, Presneau N, Odell
E, Jacques TS, Tirabosco R, Boshoff C and Flanagan AM: Brachyury, a
crucial regulator of notochordal development, is a novel biomarker
for chordomas. J Pathol. 209:157–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pillay N, Plagnol V, Tarpey PS, Lobo SB,
Presneau N, Szuhai K, Halai D, Berisha F, Cannon SR, Mead S, et al:
A common single-nucleotide variant in T is strongly associated with
chordoma. Nat Genet. 44:1185–1187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kelley MJ, Shi J, Ballew B, Hyland PL, Li
WQ, Rotunno M, Alcorta DA, Liebsch NJ, Mitchell J, Bass S, et al:
Characterization of T gene sequence variants and germline
duplications in familial and sporadic chordoma. Hum Genet.
133:1289–1297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu Z, Wang K, Wang L, Feng J, Hao S, Tian
K, Zhang L, Jia G, Wan H and Zhang J: The brachyury Gly177Asp SNP
is not associated with a risk of skull base chordoma in the Chinese
population. Int J Mol Sci. 14:21258–21265. 2013. View Article : Google Scholar : PubMed/NCBI
|