Introduction
Renal cell carcinoma (RCC) is one of the most common
malignant diseases worldwide, with 330,000 estimated new cases in
2013. Recently, an increased incidence of RCC has been observed in
developed and developing countries. Approximately 85% of RCC is
clear cell carcinoma. In the last decade, a significant improvement
in the understanding of ccRCC carcinogenesis has resulted in the
development of a predictive panel and targeted agents. Epigenetic
alteration has been shown to be an important mechanism underlying
carcinogenesis (1,2). For example, RASSF1 is a gene associated
with RCC that has been previously investigated. Epigenetic
alteration has been shown to be a dynamic process (3). In normal tissue, the methylation of
genes is different between individuals due to age, exposure and
tissue-specific origins (4). Normal
tissue may be regarded as the epigenetic background; however, the
role of elevated methylation in cancer tissue when compared with
the epigenetic background remains unknown. Pyrosequencing is a
quantitative method that allows for further elucidation (5). The identification of novel epigenetic
mechanisms underlying tumorigenesis and epigenetic dynamic
processes accompanying tumor progression is warranted.
Furthermore, epigenetic alterations could
potentially be of significant clinical value in aiding clinical
decisions. The value of epigenetic alteration in predicting Fuhrman
grade in T1 stage ccRCC has previously been reported (6); the percentage of global cellular
methylation was significantly higher in tumors of grades III and IV
(75.93 vs. 93.33%). As the management of RCC continues to evolve,
the methylation status could serve as a useful biomarker that
reflects the disease characteristics and biological behaviors.
DNA methylation arrays have recently been developed
for the genome-wide profiling of DNA methylation (7). In the current study, candidate
differentially-methylated regions (DMRs) were identified based on
this high throughput technique and were further screened in the
786-0 cell line derived from primary clear cell adenocarcinoma and
in the normal kidney tissues. DMRs that were abnormally
hypermethylated in the cell line were further validated in ccRCC
tissue and paired normal tissues by pyrosequencing. Abnormal
methylation and the associations with clinical characteristics were
examined.
Materials and methods
Methylation microarray based data
DNA methylation profiling data of 418 ccRCC samples,
consisting of 219 ccRCC and 199 paired tissues, were obtained from
The Cancer Genome Atlas project (TCGA) using the 27k Illumina
Infinium Methylation Beadchip Array (Illumina Inc., San Diego, CA,
USA) (8). Using this platform, 27,578
CpG sites in the human genome were assessed and fluorescent signals
for methylated (Cy5) and unmethylated (Cy3) alleles provided the
methylation level: β = Cy5 / (Cy3 + Cy5 + 100), with ~30 replicate
bead measurements per locus. All data were background-corrected and
normalized separately according to recommendations for each
platform. Subsequent analyses were conducted using R (a language
and environment for statistical computing and graphics) (9). Differentially-hypermethylated CpG sites
or genes between groups were identified by fold-change (fd>1.25)
and t-test (P<0.05). In the first stage, 2,856
differentially-methylated CpG sites fulfilled the P<0.05
criterion (the P-value following Bonferroni correction). In the
second stage, MBD-Seq data, which have been widely used to identify
cancer specific biomarkers (10,11), were
used to validate the results of the differentially-methylated genes
identified using the 27K methylation array, including 5 ccRCC and 5
cancer paired normal tissues.
Cell culture and
5-Aza-2′-deoxycytidine treatment of 786-0 cell line
The RCC 786-0 cell line was purchased from the Type
Culture Collection of the Chinese Academy of Sciences (Shanghai,
China) and routinely maintained in Dulbecco's modified Eagle's
medium (DMEM; Invitrogen, San Diego, CA, USA) supplemented with 10%
fetal calf serum (FCS) at 37°C in 5% CO2. Total DNA and
RNA were isolated. The demethylating agent, 5-Aza-2′-deoxycytidine
(Sigma-Aldrich, Dorset, UK), was freshly prepared in
ddH2O and filter sterilized. The cell line was plated in
75-cm2 flasks in DMEM supplemented with 10% FCS at
differing densities, depending on their doubling time, to ensure
that control and 5-Aza-2′-deoxycytidine-treated lines reached 75%
confluency at the point of RNA extraction. After 24 h, the cells
were treated with 5 mM 5-Aza-2′-deoxycytidine. The medium was
changed 24 h after treatment and then changed again after 72 h.
Following 5-Aza-2′-deoxycytidine treatment, total RNA was isolated
from the cell line. The DNA and RNA of the cell line and tissues
were isolated using the DNA/RNA Isolation kit (Tiangen Biotech Co.,
Lt., Beijing, China), according to the manufacturer's
instructions.
Tissue samples
A total of 85 paired tissue samples from patients
who underwent surgery for newly diagnosed ccRCC in Fudan University
Shanghai Cancer Center (Shanghai, China) were investigated. Each
case consisted of a sample of cancer tissue and matched paired
macroscopically normal renal tissue (distant from the tumor). The
tissues were stored at −80°C in the tissue bank. Additionally, one
case diagnosed with benign kidney angiomyolipoma was examined.
Written informed consent was obtained from the patients and the
study was approved by the Medical Institutional Review Board of
Fudan University Shanghai Cancer Center.
Bisulfite modification and methylation
analysis, and bisulfite sequencing
For bisulfite conversion, the EZ DNA
Methylation-Gold™ kit from Zymo Research Corporation (Irvine, CA,
USA) was used for optimized bisulfite conversion; 500 ng of genomic
DNA and the manufacturer's instructions were utilized. DNA was
denatured by the addition of Zymo M-Dilution buffer (containing
NaOH: Zymo Research Corporation) and incubated at 98°C for 10 min,
followed by 64°C for 2.5 h in a thermocycler. The
methylation-specific primers were designed based on the sequencing
data of the PCR-amplified bisulfite-modified cell line genome using
MethPrimer software (http://www.urogene.org/methprimer/). The DNA primers
are listed in Table I. PCR was
performed under the following conditions: Denaturation at 95°C for
5 min, followed by 42 cycles of 95°C for 30 sec, 57°C for 30 sec
and 72°C for 40 sec with a final extension step at 72°C for 10 min.
The PCR amplified fragments were cloned into pMD19-T (Takara Bio,
Inc., Otsu, Shiga, Japan) for 2 h, followed by transformation into
DH5α. A total of 8 clones were selected for PCR validation.
Transformants containing recombinant plasmids were selected by
blue/white colony screening and sequencing, as previously described
(12). The bisulfite sequencing PCR
(BSP) methylation percentage was calculated as the number of
methylated cytosines divided by the total number of cytosines in
all of the amplicons analyzed.
 | Table I.Primers. |
Table I.
Primers.
| Gene | Sequence (5′-3′) |
|---|
| BSP |
|
|
FBXW10 |
|
|
F |
TTATTTATTTTTGTTTTGGGAG |
|
R |
TTCAAAATACCAACTAAAAACC |
|
SMPD3 |
|
|
F |
TTTTTTGGTTTTAGGGTTTTGT |
|
R |
TCTCRACTTAAAACCCCAA |
| CD9 |
|
|
F |
TTTTTATAAGTGAYGTTGGGGG |
|
R |
CTAAACAATCCCCAAACRCTT |
|
HIST1H3E |
|
|
F |
TTGAAAAAATAAATTAATYGTGAA |
|
R |
AAATCCTAAACTATTTCTCRCA |
| LEP |
|
|
F |
GGAGTTGAGGATGGAGATTTAT |
|
R |
AACTCCRACRCRACTATAA |
| GGT6 |
|
|
F |
AGGAGATATTAGAGGYGTTGGT |
|
R |
ACAAACCCAACCAAACRTAAT |
| Pyrosequencing |
|
|
FBXW10 |
|
|
F |
TTGGGAGAAGTTTGTAATAGAAAAGGTA |
|
R |
Biotin-CCAAATTTTCCATAATCCTAAAATAACC |
|
Seq | AAGGTATAATTGGGG |
|
SMPD3 |
|
|
F |
GGGTGGAGGAAAGTATTGATAT |
|
R |
Biotin-CTATCCTCCTACATCCCCCTACACTAC |
|
Seq |
TGGAGGAAAGTATTGATATT |
| RT-qPCR |
|
|
FBXW10 |
|
|
F |
TCCTCCTGACTGTTAGCG |
|
R |
AACTGCACGTTGGATTGA |
|
SMPD3 |
|
|
F |
CCTTTGCGTTTCTCGGCTTTC |
|
R |
CCCGTGCCCTTCCATTCA |
Pyrosequencing
To demonstrate the relative degree of methylation,
DNA isolated from RCC and matched normal tissues were analyzed
using pyrosequencing. Measurements were performed, followed by
pyrosequencing with PyroMark Gold Q96 Reagents, using the PyroMark
Q24 pyrosequencing system (Qiagen GmbH, Hilden, Germany), according
to the manufacturer's instructions. The sequencing assay was
validated using an internal control (a non-CpG cytosine within the
target methylation sequence region). The primers are listed in
Table I.
RT-qPCR
Total RNA (1 µg) was used for cDNA synthesis using
the First Strand cDNA Synthesis kit (Fermentas, Pittsburgh, PA,
USA) following the manufacturer's protocol. RT-qPCR was performed
on PRISM 7500, (Applied Biosystems Life Technologies, Foster City,
CA, US) using SYBR Premix Ex Tag (Takara Bio Inc.). PCR was
performed under the following conditions: Denaturation at 95°C for
10 min, followed by 40 cycles of 95°C for 10 sec and 57°C for 20
sec with a final extension step at 72°C for 15 sec. Quantification
of mRNA expression was performed using the Δct method (Δct sample -
Δct calibrator). The primers are listed in Table I.
Statistical analysis
Methylation rate and the difference between RCC and
paired normal tissues (methylation rate of RCC minus that of paired
normal tissues) were analyzed.
A paired Student's t-test was used to examine
differences between the cancer and matched normal tissues.
Univariate analysis of variance or liner correlation was used to
evaluate the association between methylation status and gender,
age, Fuhrman grade, disease stage and metastasis. P<0.05 was
used to indicate a statistically significant difference in all
tests.
Results
Methylation 27K microarray data
results and locations of DMRs
At least 10 differentially-hypermethylated CpG sites
that belonged to 6 differentially-hypermethylated regions were
identified. The 6 DMRs were located in 6 genes. After searching
Pubmed, four DMRs were identified in the promoter or the first exon
of LEP, HIST1H3E, CD9 and FBXW10. Another 2 identified DMRs were
located in CpG islands belonging to the first intron of SMPD3 and
GGT6, respectively (Table II).
| Table II.Genes showing significant differential
methylation between the clear cell renall cell carcinoma and paired
adjacent tissues. |
Table II.
Genes showing significant differential
methylation between the clear cell renall cell carcinoma and paired
adjacent tissues.
| CpG name | Gene name | P-value | P-value FDR | P-value
bonferroni | Fold-change | Location |
|---|
| cg05127924 | FBXW10 |
2.11×10−73 |
5.28×10−71 |
5.28×10−69 | 1.914547176 | Exon 1 |
| cg00891541 | SMPD3 |
5.39×10−70 |
1.07×10−67 |
1.35×10−65 | 1.899386384 | Intron 1 |
| cg10556064 | SMPD3 |
2.65×10−57 |
2.73×10−55 |
6.63×10−53 | 1.682782468 | Intron 1 |
| cg19297232 | SMPD3 |
5.06×10−20 |
4.78×10−19 |
1.26×10−15 | 1.467510342 | Intron 1 |
| cg04511534 | GGT6 |
3.46×10−110 |
7.21×10−107 |
8.65×10−106 | 1.891379745 | Intron 1 |
| cg22628873 | GGT6 |
1.16×10−42 |
4.58×10−41 |
2.89×10−38 | 1.649480657 | Intron 1 |
| cg08519905 | CD9 |
7.57×10−63 |
1.06×10−60 |
1.89×10−58 | 1.906118383 | Promoter |
| cg07922606 | HIST1H3E |
2.69×10−64 |
4.15×10−62 |
6.72×10−60 | 2.009304856 | Promoter |
| cg12782180 | LEP |
1.06×10−73 |
2.69×10−71 |
2.64×10−69 | 2.208969818 | Promoter |
| cg19594666 | LEP |
2.42×10−22 |
2.63×10−21 |
6.05×10−18 | 1.550117434 | Promoter |
Methylation status of DMRs in the RCC
786-0 cell line and normal tissues
First, the candidate DMR regions were validated in
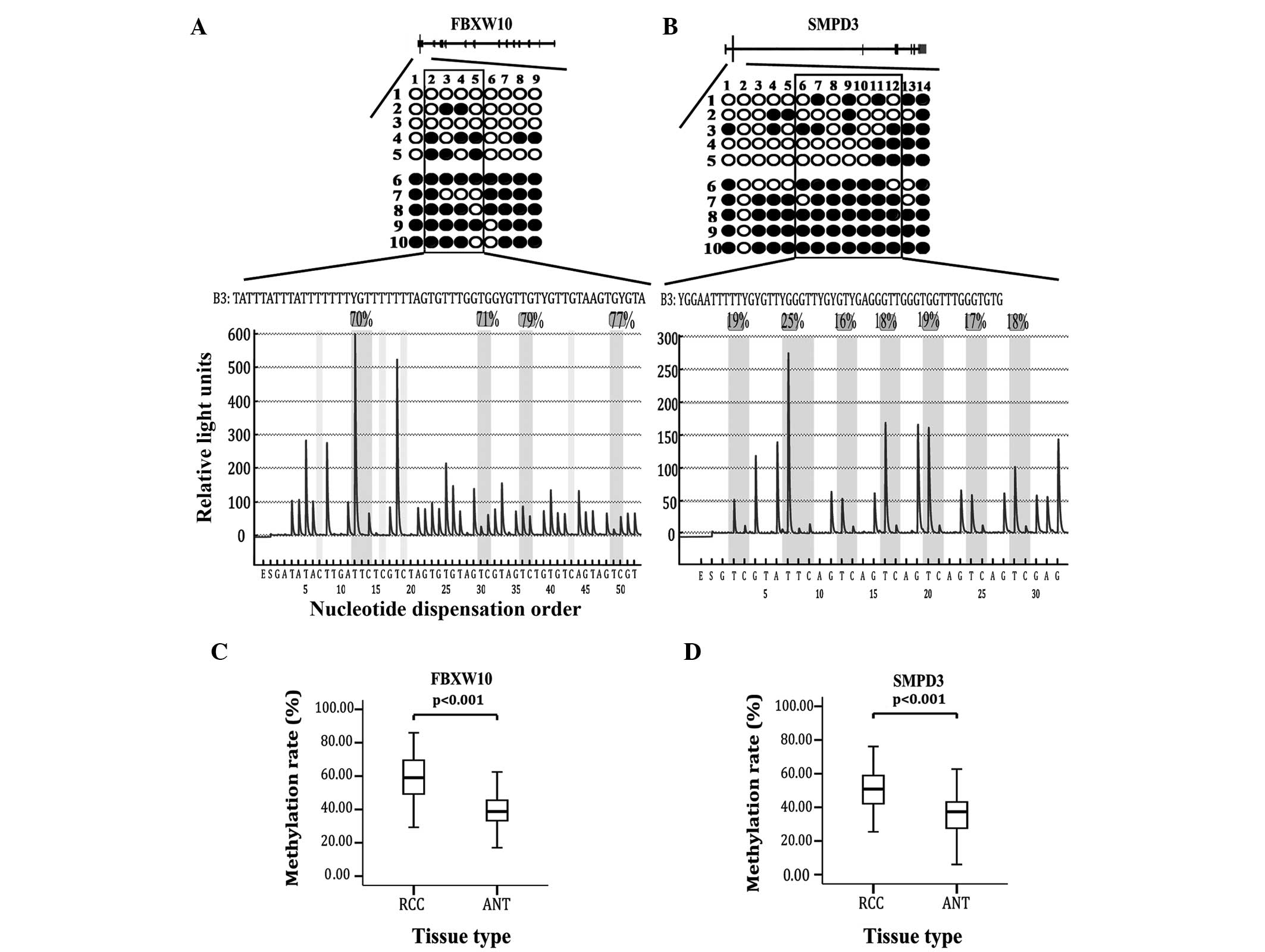
the RCC 786-0 cell line and normal renal tissues using BSP. Two
DMRs located in SMPD3 and FBXW10 were found to be hypermethylated
in the 786-0 cells only. The methylation rate of FBXW10 was 84.4%
in the 786-0 cells versus 22.2% in the normal tissues (Fig. 1A), while the methylation rate of SMPD3
was 82.9% in the 786-0 cells versus 35.7% in the normal tissues
(Fig. 1B). Additionally, 4 other DMRs
showed hypermethylation in 786-0 cells and normal tissues.
Validation of FBXW10 and SMPD3 in 85
paired RCC tissues
Further validation of the 2 hypermethylated DMRs was
performed by pyrosequencing in 85 paired RCC tissues.
Pyrosequencing of FBXW10 included 4 CpG sites (the 2nd to 5th CpG
sites in BSP), and SMPD3 included 7 CpG sites (the 6th to 12th CpG
sites in BSP) (Fig. 1A and B). The
average methylation rate of FBXW10 in the cancer tissues was
significantly higher compared with the paired normal tissues (48.78
vs. 34.62%; P<0.001; Fig. 1C).
Moreover, the methylation rate of SMPD3 was also higher in the
cancer tissues compared with the paired normal tissues (58.98 vs.
38.66%; P<0.001; Fig. 1D).
Methylation of FBXW10 and SMPD3 and
their associations with clinical characteristics in ccRCC
A total of 90.59% (77/85) cases were hypermethylated
in the RCC tissues. Upon univariate analysis, FBXW10 methylation
did not correlate with gender, age, tumor size, Fuhrman grade or
disease stage. In stage T1 disease, the methylation rate of the RCC
tissues was positively correlated with Fuhrman grade (P=0.020;
Fig. 2A). The methylation rate of
FBXW10 in Fuhrman grades I, II and III was 51.89, 60.37 and 66.18%,
respectively. Notably, the difference in methylation between the
RCC and paired normal tissues was higher in the high Fuhrman grades
(Fuhrman grade III–IV) compared with the low Fuhrman grades
(Fuhrman grade I–II). The difference in methylation rate between
the high and low Fuhrman grades was 27.24 and 21.02%, respectively
(P=0.028; Table III, Fig. 2B). Furthermore, the difference in high
stage RCC (III–IV; 31.94%) was greater than that in low stage RCC
(I–II; 22.35%) (P=0.013; Table III;
Fig. 2C).
| Table III.Clinicopathological features of
hypermethylated cases. |
Table III.
Clinicopathological features of
hypermethylated cases.
| Feature | Value | %b | P-value | %c | P-value |
|---|
| FBXW10 |
|
|
|
|
|
|
Gendera |
|
| 0.051 |
| 0.053 |
|
Male | 48 (62.34) | 63.39 |
| 25.97 |
|
|
Female | 29 (37.66) | 57.96 |
| 20.34 |
|
| Furhman
gradea |
|
| 0.162 |
| 0.028 |
|
I, II | 42 (54.55) | 59.61 |
| 21.02 |
|
|
III, IV | 35 (45.45) | 63.42 |
| 27.24 |
|
| Tumor
stagea |
|
| 0.069 |
| 0.013 |
|
I, II | 65 (84.42) | 60.28 |
| 22.35 |
|
|
III, IV | 12 (15.58) | 67.06 |
| 31.94 |
|
| Mean
age (range), years | 55 (30–84) |
| 0.388 |
| 0.344 |
| Mean
tumor size (range), cm | 6
(1.5–14) |
| 0.779 |
| 0.402 |
| SMPD3 |
|
|
|
|
|
|
Gendera |
|
| 0.113 |
| 0.786 |
|
Male | 43 (63.24) | 54.29 |
| 20.56 |
|
|
Female | 25 (36.76) | 48.55 |
| 19.55 |
|
| Furhman
gradea |
|
| 0.984 |
| 0.025 |
|
I, II | 35 (51.47) | 52.21 |
| 16.33 |
|
|
III, IV | 33 (48.53) | 52.14 |
| 24.29 |
|
| Tumor
stagea |
|
| 0.419 |
| 0.044 |
|
I, II | 57 (83.82) | 51.55 |
| 18.62 |
|
|
III, IV | 11 (16.18) | 55.42 |
| 28.38 |
|
| Mean
age (range), years | 55 (30–84) |
| 0.202 |
| 0.271 |
| Mean
tumor size (range), cm | 6
(1.5–14) |
| 0.123 |
| 0.010 |
A total of 80% (68/85) of cases were hypermethylated
in the RCC tissues. There were no significant correlations between
SMPD3 methylation rate and gender, age, tumor size, Fuhrman grade
and disease stage in the RCC tissues. Significant methylation
differences between tumor and normal tissues were found between
Fuhrman grade groups. The methylation difference was higher in the
high Fuhrman grade groups (Fuhrman grade III–IV) compared with the
low Fuhrman grade groups (Fuhrman grade I–II) (P=0.025). The
difference in methylation rate in the high and low Fuhrman grade
groups was 24.29 and 16.33%, respectively (Table III; Fig.
2D). The difference was 28.38% in the high tumor stage (III–IV)
group and 18.62% in the low tumor stage (I–II) group, which was
statistically significant (P=0.044) (Table III; Fig.
2E). There was a linear correlation between the difference in
methylation rate and the tumor size; the larger the tumor, the
greater the difference (P=0.01) (Table
III; Fig. 2F).
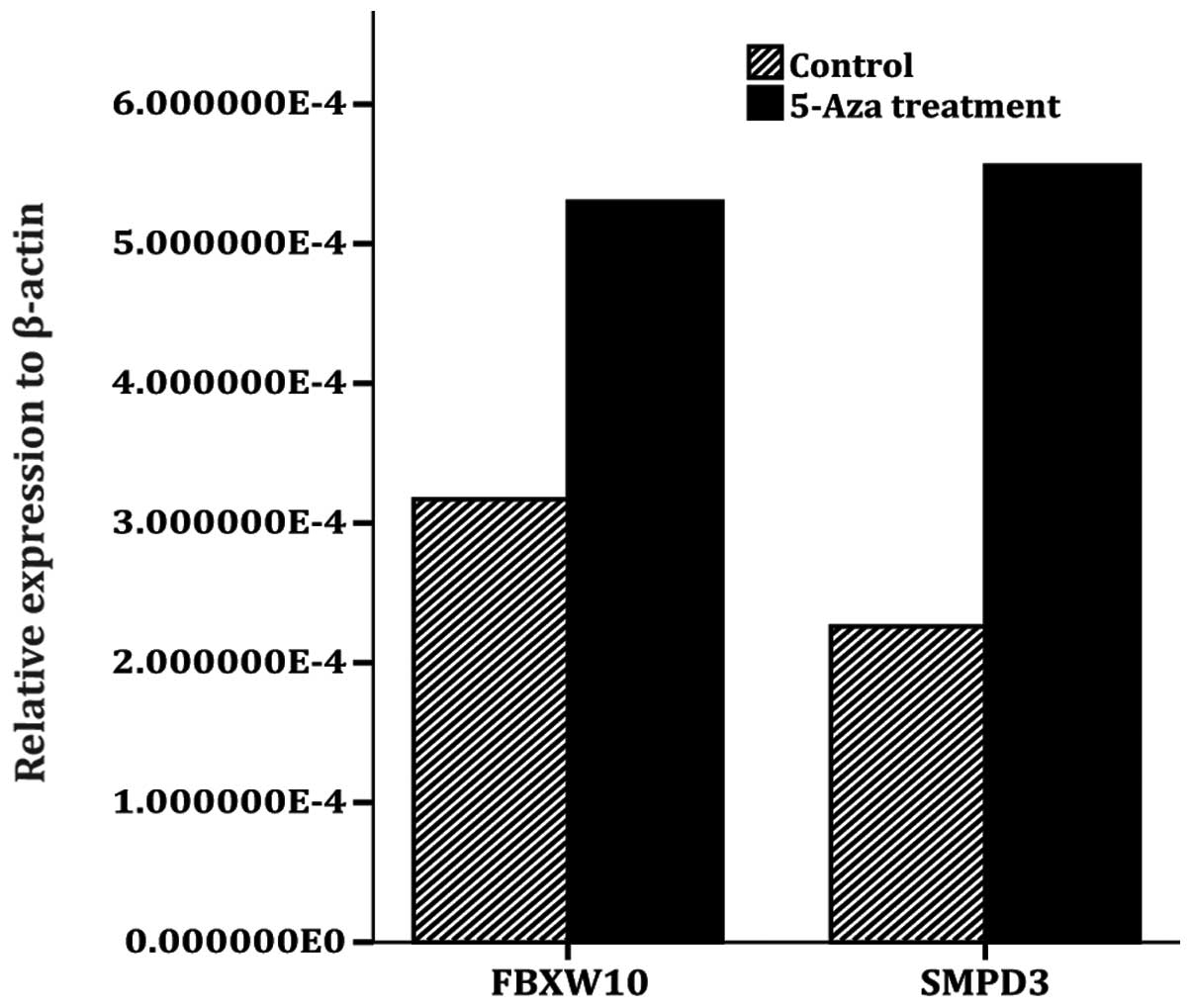
Gene expression
The RT-qPCR analysis demonstrated that SMPD3 and
FBXW10 mRNA expression was upregulated following
5-Aza-2′-deoxycytidine treatment in the RCC cell line. The
expression of SMPD3 was increased 2.47 times and that of FBXW10 was
increased 1.67 times (Fig. 3).
Discussion
The Illumina 27k methylation array platform is one
of the most comprehensive microarrays currently available for
genome-wide DNA methylation analysis. The array integrates 27,578
CpG sites (CpGs) at single-nucleotide resolution, covering
>14,000 RefSeq genes (13). The
27K methylation data from TCGA contains 418 ccRCC-associated
samples. TCGA supplies a collection of clinical and genomic data in
which associations between clinical value and genetics can be
investigated (14). In the present
study, 2,856 differentially-methylated CpG sites were identified,
and then five ccRCC and five normal tissue paired samples were used
to validate and confirm two DMRs.
FBXW10 is located in 17p12. F-box protein family
members, such as FBXW10, are characterized by an F-box motif of
~40-amino acid and act as protein-ubiquitin ligases (15). FBXW7, a member of this family that has
been widely investigated, is believed to act as a tumor suppressor
by negative regulation of a number of oncogenic proteins (16). The function of FBXW10 has not been
studied as well as methylation profiling. To the best of our
knowledge, this is the first study to report the abnormal
hypermethylation of FBXW10 in RCC. SMPD3, also known as NSMase2, is
located in 16q22.1 and is a member of the neutral sphingomyelinase
family, the major function of which is to catalyze the hydrolysis
of sphingomyelin in biological membranes to ceramide and
phosphorylcholine. Among the enzymes, SMPD3 has been the most
widely studied tumor suppressor. SMPD3 has also been implicated in
cell growth inhibition and tumorigenesis (17). The promoter of SMPD3 was identified by
hypermethylation in breast and colorectal cancer cells (18,19). The
identification was based on high throughput screening; however, the
methylation status in ccRCC has not been reported to date.
The DMR of FBXW10 is located in the head of the
first exon. Hypermethylation has been previously demonstrated in
cell lines and tumor tissues (20).
As the first exon is a common locus that causes epigenetic
inactivation, the present study analyzed further gene expression
regulated by methylation using demethylation treatment. The
upregulation of expression following 5-Aza-2′-deoxycytidine
treatment indicated that FBXW10 exhibited epigenetic inactivation
by hypermethylation of the first exon. The DMR of SMPD3 was located
in the head of the first intron within 1 kb downstream of the first
exon. As a tumor suppressor that has been widely studied, the
promoter of SMPD3 was identified by hypermethylation in breast and
colorectal cancer cells (17). In the
present study, a novel hypermethylated DMR was identified in the
ccRCC cells and tissues. Although it is not a traditional region of
inactivated gene expression, further demethylation treatment caused
upregulation of expression. Previous studies have shown that the
hypermethylation of introns also regulates gene expression
(21,22). The underlying mechanisms may involve
binding proteins that regulate methylation and methylation of the
first intron of SMPD3, thus affecting gene expression (21). In the present study, it was reported
for the first time that novel hypermethylation is another potential
mechanism that downregulates gene expression, with the exception of
promoter methylation in SMPD3.
The methylation rate of FBXW10 correlated with the
Fuhrman grade in T1 stage ccRCC. The higher the methylation rate,
the higher the Fuhrman grade. Due to the unsatisfactory accuracy of
the Fuhrman grade and percutaneous renal biopsy, the methylation
rate of FBXW10 could serve as a beneficial biomarker to improve the
accuracy of diagnosis. T1 stage ccRCC with FBXW10 hypermethylation
tends to be highly invasive. Thus, this additional tool could aid
in the identification of the optimal treatment of T1 stage ccRCC
(23–25). A previous study showed that FBXW10
acted as a protein-ubiquitin ligase. F-box proteins interact with
SKP1 via F box, and interact with ubiquitination targets through
other protein interaction domains (15). The present study is the first to
identify methylation profiling as a biomarker; it was demonstrated
that FBXW10 was inactivated by methylation of the first exon, and
that inactivation of FBXW10 may be significant in the formation of
a highly invasive tumor. The paired normal tissue chosen in this
study was normal renal cortex tissue distant from the tumor. As the
methylation of genes is different in individuals due to age,
exposure and tissue-specific origins, the methylation status of the
paired normal tissue could be regarded as the epigenetic background
of the patient. In the present study, the difference was defined as
the methylation rate of RCC minus the methylation rate of the
paired normal tissue. This enabled investigation of the dynamic
process of DNA methylation. The results indicated that not only the
RCC methylation rate was important, but that it also reflected the
difference between participants with high Fuhrman grade formation
and progression of local tumor. This is the first study to
demonstrate the link between cancer and individualized epigenetic
background. However, the function of FBXW10 in ccRCC requires
further elucidation.
The correlation of the SMPD3 methylation rate with
clinical features also exhibited differences. Moreover, the results
indicated differences in the participants with high Fuhrman grade
formation and tumor progression. Linear correlation analysis
indicated that the larger the tumor, the higher the methylation
rate in RCC compared to the background. This dynamic process
contributes to tumor growth.
The present study was not without limitations. All
tumor stages were selected to obtain a general sense of methylation
profiling. Larger samples could be studied to confirm the value of
FBXW10 as a biomarker. Further studies could reveal the function of
methylation regulation of the two novel genes.
In conclusion, two novel DMRs located in SMPD3 and
FBXW10 were found to be hypermethylated in ccRCC tissues, and
participants exhibited hypermethylated inactivation and tumor
genes. FBXW10 was a useful biomarker to predict Fuhrman grade in T1
stage tumors and improved the accuracy of percutaneous renal
biopsy. The findings suggest that it is not the methylation rate of
RCC, but rather the dynamic methylation process that is important
in participants with high Fuhrman grade formation and tumor
stage.
Glossary
Abbreviations
Abbreviations:
|
ccRCC
|
clear cell renal cell carcinoma
|
|
DMR
|
differentially-methylated regions
|
|
RT-qPCR
|
reverse transcription quantitative
polymerase chain reaction
|
|
TCGA
|
The Cancer Genome Atlas project
|
References
|
1
|
Morrissey C, Martinez A, Zatyka M, et al:
Epigenetic inactivation of the RASSF1A 3p21.3 tumor suppressor gene
in both clear cell and papillary renal cell carcinoma. Cancer Res.
61:7277–7281. 2001.PubMed/NCBI
|
|
2
|
Kawai Y, Sakano S, Suehiro Y, et al:
Methylation level of the RASSF1A promoter is an independent
prognostic factor for clear-cell renal cell carcinoma. Ann Oncol.
21:1612–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome-biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Christensen BC, Houseman EA, Marsit CJ, et
al: Aging and environmental exposures alter tissue-specific DNA
methylation dependent upon CpG island context. PLoS Genet.
5:e10006022009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gharizadeh B, Nordström T, Ahmadian A, et
al: Long-read pyrosequencing using pure
2′-deoxyadenosine-5′-O'-(1-thiotriphosphate) Sp-isomer. Anal
Biochem. 301:82–90. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Minardi D, Lucarini G, Filosa A, et al:
Prognostic role of global DNA-methylation and histone acetylation
in pT1a clear cell renal carcinoma in partial nephrectomy
specimens. J Cell Mol Med. 13:2115–2121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Houseman EA, Accomando WP, Koestler DC, et
al: DNA methylation arrays as surrogate measures of cell mixture
distribution. BMC Bioinformatics. 13:862012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bibikova M, Le J, Barnes B, et al:
Genome-wide DNA methylation profiling using Infinium® assay.
Epigenomics. 1:177–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dessau RB and Pipper CB: ‘R’-project for
statistical computing. Ugeskr Laeger. 170:328–330. 2008.(In
Danish). PubMed/NCBI
|
|
10
|
Zhao Y, Guo S, Sun J, et al: Methylcap-seq
reveals novel DNA methylation markers for the diagnosis and
recurrence prediction of bladder cancer in a Chinese population.
PLoS One. 7:e351752012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He Y, Cui Y, Wang W, et al:
Hypomethylation of the hsa-miR-191 locus causes high expression of
hsa-mir-191 and promotes the epithelial-to-mesenchymal transition
in hepatocellular carcinoma. Neoplasia. 13:841–853. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu J, Zhu T, Wang Z, et al: A novel set of
DNA methylation markers in urine sediments for sensitive/specific
detection of bladder cancer. Clin Cancer Res. 13:7296–7304. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen YA, Choufani S, Ferreira JC,
Grafodatskaya D, Butcher DT and Weksberg R: Sequence overlap
between autosomal and sex-linked probes on the Illumina
HumanMethylation27 microarray. Genomics. 97:214–222. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Braun R, Finney R, Yan C, et al: Discovery
analysis of TCGA data reveals association between germline genotype
and survival in ovarian cancer patients. PLoS One. 8:e550372013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jin J, Cardozo T, Lovering RC, Elledge SJ,
Pagano M and Harper JW: Systematic analysis and nomenclature of
mammalian F-box proteins. Genes Dev. 18:2573–2580. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou ZY, Tu KS, Zhang J, et al: Expression
of Fbxw7 and its correlation with cell proliferation in human
hepatocellular carcinoma. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi.
28:1303–1306. 2012.(In Chinese). PubMed/NCBI
|
|
17
|
Clarke CJ, Guthrie JM and Hannun YA:
Regulation of neutral sphingomyelinase-2 (nSMase2) by tumor
necrosis factor-alpha involves protein kinase C-delta in lung
epithelial cells. Mol Pharmacol. 74:1022–1032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Demircan B, Dyer LM, Gerace M, Lobenhofer
EK, Robertson KD and Brown KD: Comparative epigenomics of human and
mouse mammary tumors. Genes Chromosomes Cancer. 48:83–97. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shen Y, Takahashi M, Byun HM, et al:
Boswellic acid induces epigenetic alterations by modulating DNA
methylation in colorectal cancer cells. Cancer Biol Ther.
13:542–552. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chaturvedi P and Parnaik VK: Lamin A rod
domain mutants target heterochromatin protein 1alpha and beta for
proteasomal degradation by activation of F-box protein, FBXW10.
PLoS One. 5:e106202010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang Y, Liu C, Wang X, Liu D, Ingvarsson S
and Chen H: Demethylation of the region around exon 2 of MLH1 gene
in gastrointestinal cancer. Anticancer Res. 32:4861–4864.
2012.PubMed/NCBI
|
|
22
|
Godler DE, Slater HR, Bui QM, et al: FMR1
intron 1 methylation predicts FMRP expression in blood of female
carriers of expanded FMR1 alleles. J Mol Diagn. 13:528–536. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smaldone MC and Uzzo RG: Active
surveillance: a potential strategy for select patients with small
renal masses. Future Oncol. 7:1133–1147. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lane BR, Tobert CM and Riedinger CB:
Growth kinetics and active surveillance for small renal masses.
Curr Opin Urol. 22:353–359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mason RJ, Abdolell M, Trottier G, et al:
Growth kinetics of renal masses: analysis of a prospective cohort
of patients undergoing active surveillance. Eur Urol. 59:863–867.
2011. View Article : Google Scholar : PubMed/NCBI
|