Introduction
Neuroblastoma is the most frequent extracranial
solid tumor in children and is characterized by its extreme
heterogeneity, ranging from spontaneous regression to malignant
progression. More than half of neuroblastoma patients are
stratified into a high-risk group and <40% of these high-risk
patients can expect long-term survival. This is mainly due to the
chemoresistant minimal residual disease (MRD) that is primarily
responsible for tumor metastasis and relapse (1–3).
Although tumor cell dissemination is traditionally
classified as a late event during tumor progression, accumulating
evidence suggests that tumor cells disseminate from the primary
lesions even before the formation of overt tumors, and become
circulating tumor cells (CTCs) in the peripheral blood (PB) and
disseminating tumor cells (DTCs) in the bone marrow (BM) (4–6). Following
local and systemic treatment, residual tumor cells remain as CTCs
in the PB and DTCs in the BM, as well as cancer stem cells in the
primary lesions. Due to the extremely restricted availability of
primary tumor samples, PB and BM samples are mainly used for MRD
monitoring in the clinics (7–9).
As sensitive detection of MRD is essential for
monitoring disease status and evaluating treatment response in
high-risk neuroblastoma patients, a number of MRD detection
protocols based on reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) markers have been reported (10–13).
Although the ideal markers should be exclusively expressed in
neuroblastoma cells, the currently available markers are selected
by their ability to define a cut-off value that distinguishes
neuroblastoma cells from normal PB and BM cells.
To overcome this limitation, the current protocols
utilize multiple MRD markers for PB and BM samples, which are
either common or distinct. Common MRD markers are reported as
three-marker [double-cortin (DCX), paired-like homeobox 2b (PHOX2B)
and tyrosine hydroxylase (TH)] and eight-marker [(cyclin D1,
collapsin response mediator protein 1 (CRMP1), dopa decarboxylase
(DDC), GABA A receptor β3 (GABRB3), ISL LIM homeobox 1 (ISL1),
kinesin family member 1A (KIF1A), PHOX2B and transforming acidic
coiled-coil-containing protein 2] sets (10,11), while
distinct MRD markers are reported as the PB set [PHOX2B, TH, DDC,
dopamine β-hydroxylase (DBH) and cholinergic receptor, nicotinic,
α3 (CHRNA3)] and BM set [(PHOX2B, TH, DDC, CHRNA3 and
growth-associated protein 43 (GAP43)] (12). However, the rationale for introducing
the current protocols into the clinics remains unclear (14–16).
In the present study, we determined the expression
of 11 previously validated MRD markers (CHRNA3, CRMP1, DBH, DCX,
DDC, GABRB3, GAP43, ISL1, KIF1A, PHOX2B and TH) in 23 pairs of PB
and BM samples collected from seven high-risk neuroblastoma
patients treated at Kobe University Hospital and Kobe Children's
Hospital, Japan, between November 2011 and April 2014 (13), and analyzed the correlation between PB
and BM samples.
Materials and methods
Patients and samples
All PB and BM samples were obtained from seven
high-risk neuroblastoma patients with written informed consent. All
patients were treated at Kobe University Hospital and Kobe
Children's Hospital between November 2011 and April 2014. The use
of human samples for this study was approved by the Ethics
Committee at Kobe University Graduate School of Medicine and
conducted in accordance with the Guidelines for the Clinical
Research of Kobe University Graduate School of Medicine.
RNA extraction and cDNA synthesis
All PB and BM samples were separated using Mono-Poly
resolving medium (DS Pharma Biomedical, Osaka, Japan), and
nucleated cells were collected according to the manufacturer's
instructions. Total RNA was then extracted with a TRIzol Plus RNA
purification kit (Life Technologies, Carlsbad, CA, USA) according
to the manufacturer's instructions. After evaluating RNA integrity
by agarose gel electrophoresis, cDNA was synthesized from 1 or 0.5
µg total RNA using a Quantitect reverse transcription kit (Qiagen,
Valencia, CA, USA) and diluted to a total volume of 80 or 40
µl.
RT-qPCR
RT-qPCR was performed using an ABI 7500 Fast
real-time PCR system (Applied Biosystems, Foster City, CA, USA) in
a total volume of 15 µl consisting of 7.5 µl 2X FastStart Universal
SYBR-Green Master (Roche, Mannheim, Germany), 1.5 µl each of 3 µM
sense and anti-sense primers, and 1 µl sample cDNA (corresponding
to 12.5 ng total RNA). Each cDNA was amplified with a precycling
hold at 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec
and 60°C for 60 sec, and one cycle at 95°C for 15 sec, 60°C for 60
sec, 95°C for 15 sec, and 60°C for 15 sec. Each sample was analyzed
in triplicate. The expression of the 11 MRD markers (CHRNA3, CRMP1,
DBH, DCX, DDC, GABRB3, GAP43, ISL1, KIF1A, PHOX2B and TH) was
calculated based on the relative standard curve method using
β2-microglobulin as an endogenous reference for normalization, and
was scored as positive if its expression exceeded the normal range
(13).
Statistical analysis
Differences between the number of MRD-positive
samples in PB and BM were evaluated by McNemar's Chi-squared test.
To assess the correlation between MRD marker expression in PB and
BM samples, the expression of each marker was ranked according to
the number of positive samples in 23 PB and 23 BM samples.
Correlation between the rank in PB and BM samples was assessed by
Spearman's rank correlation coefficient. P<0.05 was considered
to indicate a statistically significant difference. Statistical
analyses were performed with EZR (version 1.24 www.jichi.ac.jp/saitama-sct/SaitamaHP.files/statmedEN.html;
Saitama Medical Centre, Jichi Medical University, Saitama, Japan)
(17).
Results
Characteristics of PB and BM
samples
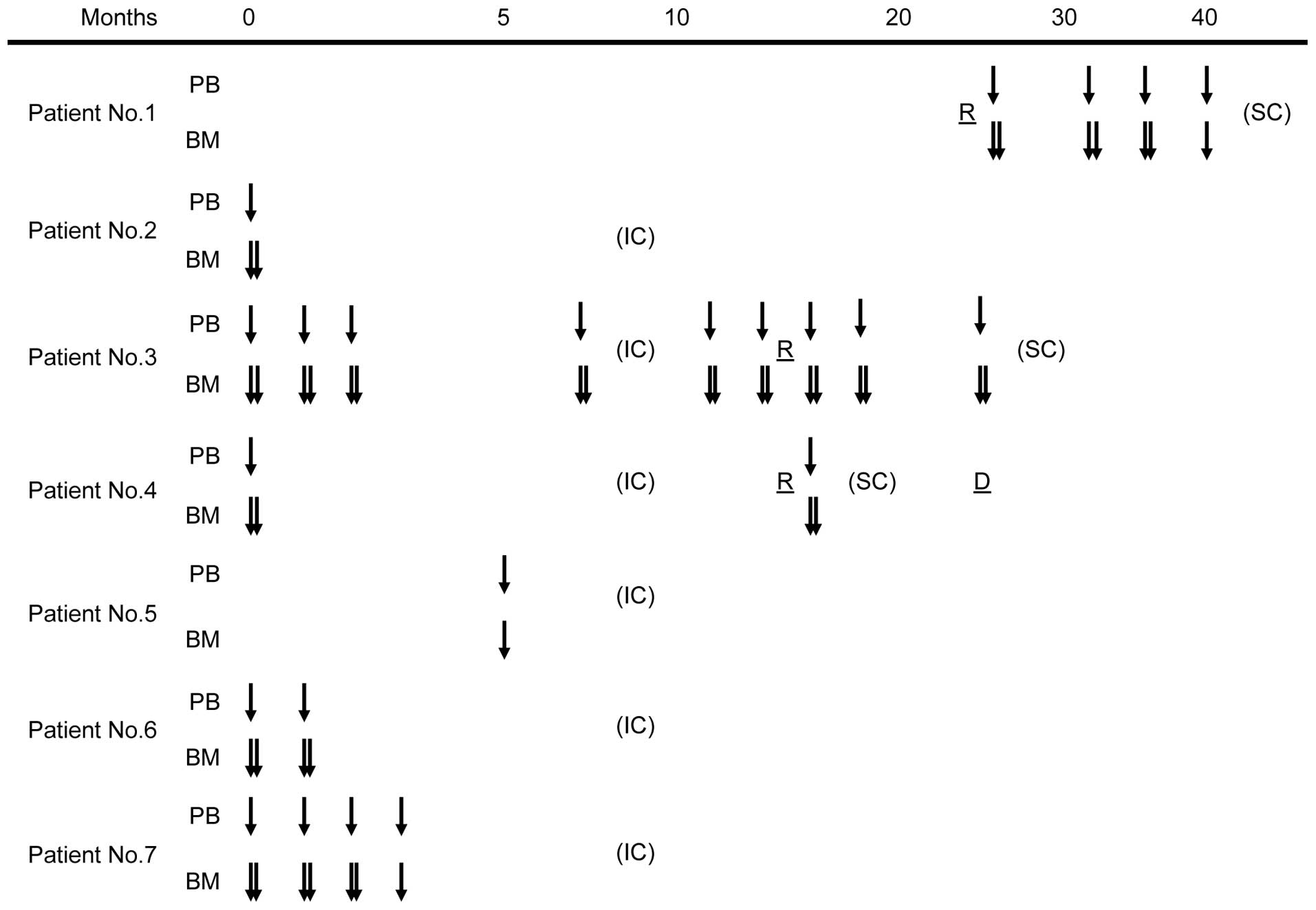
The 23 pairs of PB and BM samples were obtained at
the same time point from seven high-risk neuroblastoma patients who
were treated at Kobe University Hospital and Kobe Children's
Hospital between November 2011 and April 2014 (Fig. 1). Patient characteristics are shown in
Table I. All patients were stratified
into the high-risk group (18) and
treated with induction chemotherapy followed by peripheral blood
stem cell transplantation, radiation therapy and surgical therapy
according to the Japan Neuroblastoma Study Group protocol. Patients
1, 3 and 4 experienced tumor relapse and underwent salvage
chemotherapy. The median follow-up time was 24 months (range, 7–29
months).
 | Table I.Patient characteristics. |
Table I.
Patient characteristics.
| Patient number | Age | Gender | Tumor origin | INSS stage | MYCN status | Follow-up | Present status |
|---|
| 1 | 3 y | M |
Adrenal gland | 4 |
Non-amplified | 25–49 m |
Alive (Relapsed) |
| 2 | 4 y | M | Adrenal gland | 3 | Non-amplified | 0–29 m | Alive
(Relapse-free) |
| 3 | 2 y | M | Adrenal gland | 4 | Amplified | 0–24 m | Alive (Relapsed) |
| 4 | 3 y | F | Adrenal gland | 4 | Amplified | 0–24 m | Deceased
(Relapsed) |
| 5 | 5 y | M | Posterior
mediastinum | 4 | Non-amplified | 0–17 m | Alive
(Relapse-free) |
| 6 | 11 m | M | Adrenal gland | 4 | Amplified | 0–9 m | Alive
(Relapse-free) |
| 7 | 14 m | M | Adrenal gland | 4 | Amplified | 0–7 m | Alive
(Relapse-free) |
CHRNA3, CRMP1, DBH, DCX, DDC, GABRB3, GAP43, ISL1,
KIF1A, PHOX2B and TH expression was determined by RT-qPCR, and was
scored as positive if its expression exceeded the normal range
(13). The number of positive MRD
markers in each sample is presented in Table II. A sample was scored as
MRD-positive if it had more than one positive marker. There was no
statistically significant difference between the number of
MRD-positive samples in PB and BM samples (Table III, P=1.000).
| Table II.Sample characteristics. |
Table II.
Sample characteristics.
|
| Number of positive
markers |
|---|
|
|
|
|---|
| Sample pair
number | PB sample | BM sample |
|---|
| 1 | 1 | 3 |
| 2 | 2 | 10 |
| 3 | 0 | 9 |
| 4 | 0 | 0 |
| 5 | 1 | 11 |
| 6 | 1 | 1 |
| 7 | 1 | 0 |
| 8 | 0 | 0 |
| 9 | 1 | 0 |
| 10 | 0 | 1 |
| 11 | 1 | 0 |
| 12 | 2 | 11 |
| 13 | 6 | 11 |
| 14 | 2 | 11 |
| 15 | 1 | 1 |
| 16 | 10 | 11 |
| 17 | 0 | 1 |
| 18 | 0 | 1 |
| 19 | 2 | 1 |
| 20 | 0 | 10 |
| 21 | 1 | 7 |
| 22 | 1 | 0 |
| 23 | 0 | 0 |
| Table III.MRD monitoring in PB and BM
samples. |
Table III.
MRD monitoring in PB and BM
samples.
|
| BM sample |
|---|
|
|
|
|---|
| PB sample | MRD (+) | MRD (−) |
|---|
| MRD (+) | 11 | 4 |
| MRD (−) | 5 | 3 |
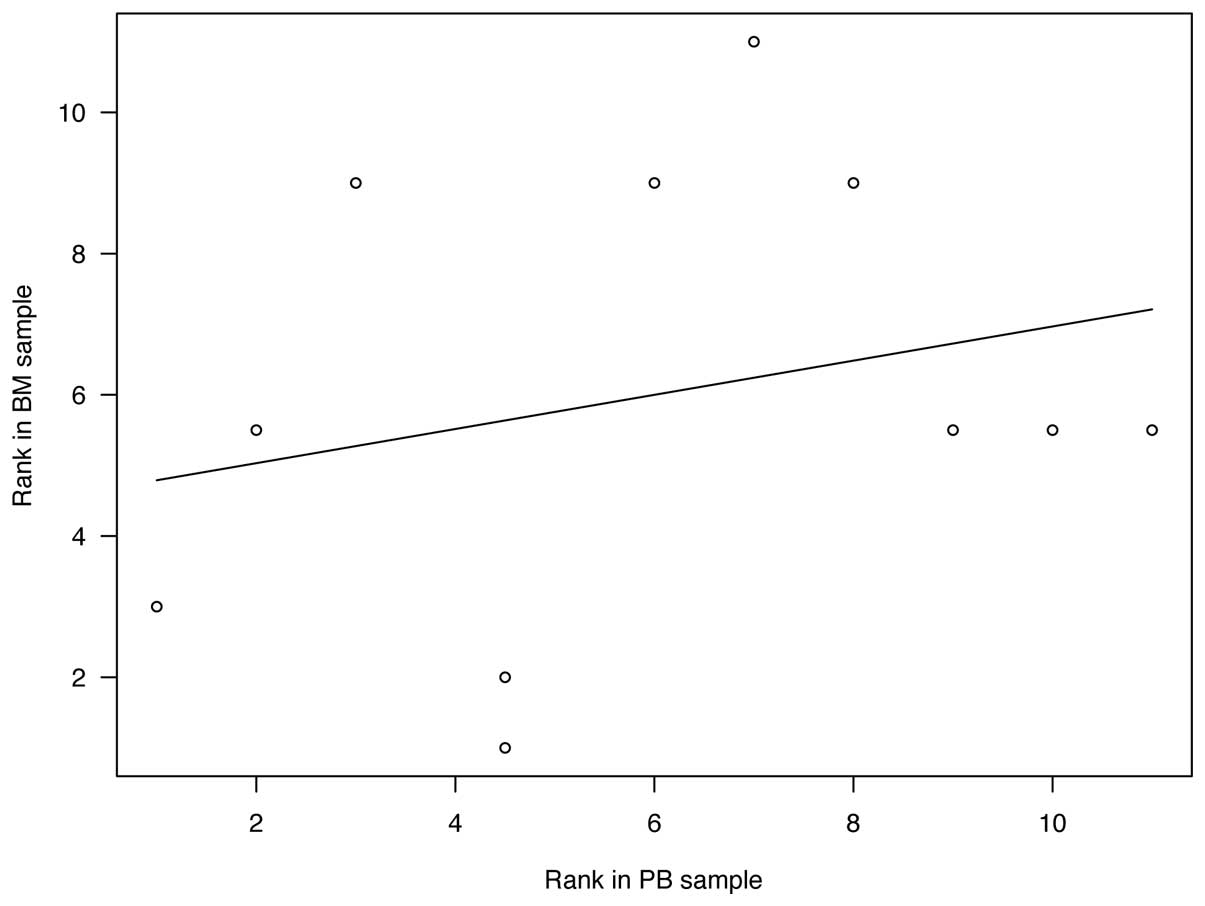
Correlation between MRD marker
expression in PB and BM samples
The number of positive samples of each MRD marker in
PB and BM samples is shown in Table
IV. CRMP1 and KIF1A were ranked as the two most sensitive
markers for PB samples, whereas these were PHOX2B and DBH for BM
samples. There was no statistical significance in the correlation
between the rank of MRD markers in PB and BM samples (Fig. 2, r=0.250, P=0.459).
| Table IV.MRD marker expression in PB and BM
samples. |
Table IV.
MRD marker expression in PB and BM
samples.
| MRD marker | PB sample | BM sample |
|---|
| CHRNA3 |
|
|
|
(+) | 1 | 9 |
|
(−) | 20 | 14 |
| CRMP1 |
|
|
|
(+) | 9 | 10 |
|
(−) | 14 | 13 |
| DBH |
|
|
|
(+) | 3 | 12 |
|
(−) | 20 | 11 |
| DCX |
|
|
|
(+) | 4 | 8 |
|
(−) | 19 | 15 |
| DDC |
|
|
|
(+) | 1 | 9 |
|
(−) | 22 | 14 |
| GABRB3 |
|
|
|
(+) | 2 | 5 |
|
(−) | 21 | 18 |
| GAP43 |
|
|
|
(+) | 2 | 8 |
|
(−) | 19 | 15 |
| ISL1 |
|
|
|
(+) | 1 | 8 |
|
(−) | 19 | 15 |
| KIF1A |
|
|
|
(+) | 6 | 9 |
|
(−) | 17 | 14 |
| PHOX2B |
|
|
|
(+) | 3 | 13 |
|
(−) | 20 | 10 |
| TH |
|
|
|
(+) | 1 | 9 |
|
(−) | 22 | 14 |
Discussion
To improve the outcome of high-risk neuroblastoma
patients, sensitive MRD detection is essential for evaluating the
disease status and treatment response. Although MRD may be detected
in PB as well as BM samples, the correlation of MRD marker
expression between the PB and BM samples remains elusive. In the
present study, we determined the expression of 11 previously
validated MRD markers (CHRNA3, CRMP1, DBH, DCX, DDC, GABRB3, GAP43,
ISL1, KIF1A, PHOX2B and TH) in 23 pairs of PB and BM samples
obtained from seven high-risk neuroblastoma patients treated at
Kobe University Hospital and Kobe Children's Hospital between
November 2011 and April 2014 (13).
Although the number of MRD-positive samples was not significantly
different between PB and BM samples, there was no significant
correlation between the expression of these markers in the
samples.
In the present study, we collected the 23 pairs of
PB and BM samples from the same patient at the same time point in
order to minimize the variability of MRD marker expression
(19). Even under these conditions,
the sensitivity of MRD markers in PB samples was clearly different
from that in BM samples (Table IV).
KIF1A and DCX were the only positive markers in PB but not BM
samples, whereas PHOX2B and DBH were positive in BM but not PB
samples. Although DBH was previously listed as an MRD marker for PB
samples (12), the present study
identified it as being one of the most sensitive markers in BM
samples. As suggested for anti-GD2 antibody and
metaiodobenzylguanidine (MIBG) therapies (11), the various treatment protocols might
affect these inconsistencies.
Although the quantity of MRD in PB and/or BM samples
predicts tumor relapse and patient outcome, conflicting results
have been reported with regard to the prognostic value of MRD
monitoring using various MRD markers (14–16). Given
that CTCs in the PB and DTCs in the BM define the main faces of MRD
in the clinics, these inconsistencies may imply genetic and
phenotypic heterogeneity of CTCs and DTCs (20–22).
Although CTCs have not been convincingly isolated from
neuroblastoma patients, as demonstrated in breast and lung cancers
(23,24), the present results reveal the need for
careful selection of MRD markers for PB and BM samples.
In summary, the expression of 11 previously
validated MRD markers in PB and BM samples from high-risk
neuroblastoma patients was not significantly correlated. Distinct
markers for PB and BM samples may be required to achieve sensitive
MRD detection in neuroblastoma patients.
Acknowledgements
This study was supported in part by Grants-in-Aid
for Scientific Research from the Ministry of Education, Culture,
Sports, Science and Technology of Japan, and grants from the
Children's Cancer Association of Japan and Hyogo Science and
Technology Association.
References
|
1
|
Brodeur GM: Neuroblastoma: Biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maris JM: Recent advances in
neuroblastoma. N Engl J Med. 362:2202–2211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hüsemann Y, Geigl JB, Schubert F, Musiani
P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmüller G and
Klein CA: Systemic spread is an early step in breast cancer. Cancer
Cell. 13:58–68. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rhim AD, Mirek ET, Aiello NM, et al: EMT
and dissemination precede pancreatic tumor formation. Cell.
148:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kang Y and Pantel K: Tumor cell
dissemination: Emerging biological insights from animal models and
cancer patients. Cancer Cell. 23:573–581. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Müller V, Alix-Panabières C and Pantel K:
Insights into minimal residual disease in cancer patients:
Implications for anti-cancer therapies. Eur J Cancer. 46:1189–1197.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin H, Balic M, Zheng S, Datar R and Cote
RJ: Disseminated and circulating tumor cells: Role in effective
cancer management. Crit Rev Oncol Hematol. 77:1–11. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mordant P, Loriot Y, Lahon B, Castier Y,
Lesèche G, Soria JC, Massard C and Deutsch E: Minimal residual
disease in solid neoplasia: New frontier or red-herring? Cancer
Treat Rev. 38:101–110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Viprey VF, Lastowska MA, Corrias MV,
Swerts K, Jackson MS and Burchill SA: Minimal disease monitoring by
QRT-PCR: guidelines for identification and systematic validation of
molecular markers prior to evaluation in prospective clinical
trials. J Pathol. 216:245–252. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheung IY, Feng Y, Gerald W and Cheung NK:
Exploiting gene expression profiling to identify novel minimal
residual disease markers of neuroblastoma. Clin Cancer Res.
14:7020–7027. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stutterheim J, Gerritsen A,
Zappeij-Kannegieter L, Yalcin B, Dee R, van Noesel MM, Berthold F,
Versteeg R, Caron HN, van der Schoot CE and Tytgat GA: Detecting
minimal residual disease in neuroblastoma: The superiority of a
panel of real-time quantitative PCR markers. Clin Chem.
55:1316–1326. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hartomo TB, Kozaki A, Hasegawa D, Van
Huyen Pham T, Yamamoto N, Saitoh A, Ishida T, Kawasaki K, Kosaka Y,
Ohashi H, et al: Minimal residual disease monitoring in
neuroblastoma patients based on the expression of a set of
real-time RT-PCR markers in tumor-initiating cells. Oncol Rep.
29:1629–1636. 2013.PubMed/NCBI
|
|
14
|
Stutterheim J, Zappeij-Kannegieter L,
Versteeg R, Caron HN, van der Schoot CE and Tytgat GA: The
prognostic value of fast molecular response of marrow disease in
patients aged over 1 year with stage 4 neuroblastoma. Eur J Cancer.
47:1193–1202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yáñez Y, Grau E, Oltra S, Cañete A,
Martínez F, Orellana C, Noguera R, Palanca S and Castel V: Minimal
disease detection in peripheral blood and bone marrow from patients
with non-metastatic neuroblastoma. J Cancer Res Clin Oncol.
137:1263–1272. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Corrias MV, Haupt R, Carlini B, Cappelli
E, Giardino S, Tripodi G, Tonini GP, Garaventa A, Pistoia V and
Pistorio A: Multiple target molecular monitoring of bone marrow and
peripheral blood samples from patients with localized neuroblastoma
and healthy donors. Pediatr Blood Cancer. 58:43–49. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kanda Y: Investigation of the freely
available easy-to-use software ‘EZR’ for medical statistics. Bone
Marrow Transplant. 48:452–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Castleberry RP, Pritchard J, Ambros P,
Berthold F, Brodeur GM, Castel V, Cohn SL, De Bernardi B,
Dicks-Mireaux C, Frappaz D, et al: The international neuroblastoma
risk groups (INRG): a preliminary report. Eur J Cancer.
33:2113–2116. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stutterheim J, Zappeij-Kannegieter L, Ora
I, van Sluis PG, Bras J, den Ouden E, Versteeg R, Caron HN, van der
Schoot CE and Tytgat GA: Stability of PCR targets for monitoring
minimal residual disease in neuroblastoma. J Mol Diagn. 14:168–175.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vermeulen L, de Sousa e Melo F, Richel DJ
and Medema JP: The developing cancer stem-cell model: clinical
challenges and opportunities. Lancet Oncol. 13:e83–e89. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Plaks V, Koopman CD and Werb Z: Cancer:
Circulating tumor cells. Science. 341:1186–1188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alix-Panabières C and Pantel K: Challenges
in circulating tumour cell research. Nat Rev Cancer. 14:623–631.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baccelli I, Schneeweiss A, Riethdorf S,
Stenzinger A, Schillert A, Vogel V, Klein C, Saini M, Bäuerle T,
Wallwiener M, et al: Identification of a population of blood
circulating tumor cells from breast cancer patients that initiates
metastasis in a xenograft assay. Nat Biotechnol. 31:539–544. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hodgkinson CL, Morrow CJ, Li Y, Metcalf
RL, Rothwell DG, Trapani F, Polanski R, Burt DJ, Simpson KL, Morris
K, et al: Tumorigenicity and genetic profiling of circulating tumor
cells in small-cell lung cancer. Nat Med. 20:897–903. 2014.
View Article : Google Scholar : PubMed/NCBI
|