Introduction
A major challenge in the field of breast cancer is
the identification and exploitation of useful therapeutic targets
for the most aggressive forms. Among these, the triple-negative
breast cancers (TNBCs), characterized by a lack of estrogen,
progesterone and human epidermal growth factor receptor-2 (HER2)
receptors, are a highly heterogeneous group of tumors which account
for ~15% of all breast cancers. TNBCs are often associated with
epithelial-mesenchymal transition and a high propensity for early
metastasis (1). To date, no
molecularly-targeted therapeutic agents are clinically available
for TNBCs, therefore these tumors, which are frequently resistant
to cytotoxic chemotherapy, remain challenging to treat.
Nevertheless, progress is being made in the subtyping of TNBCs
following the identification of molecular alterations, which
current therapeutic efforts are focused towards (2).
With regard to the identification of molecular
alterations in TNBCs, various studies have indicated the potential
importance of the dysregulation of the Raf-1 kinase inhibitor
protein (RKIP) signaling pathway in breast cancer metastasis and
the biology of TNBCs (3–5). RKIP inhibits the Raf-1/mitogen-activated
protein kinase kinase interaction and thus the oncogenic activities
of the mitogen activated protein kinase pathway (6). RKIP also affects the activation of
nuclear factor-κB (NF-κB) and signal transducer and activator of
transcription-3 (7,8), antagonizing their pro-oncogenic effects.
In its phosphorylated state, RKIP downregulates numerous G
protein-coupled receptors and may also be involved in regulation of
the partitioning of chromosomes and progression through mitosis
(9,10). Overall, RKIP is a tumor and metastasis
suppressor, which is downregulated in numerous types of cancer,
including breast cancer, and is correlated with a more severe
prognosis (11). Furthermore, RKIP is
able to promote drug-induced apoptosis in cancer cells (12), underscoring the need for novel
approaches for the rescue of RKIP expression levels in neoplastic
tissues.
Unfortunately, the causes underlying RKIP
downregulation in these tumors remain to be elucidated. In the case
of breast cancer, the reduction in RKIP has been associated with
transcriptional repression by the NF-κB/Snail pathway (13,14) or the
polycomb protein enhancer of zeste homolog 2 (15), as well as silencing by microRNA-224
(miR-224) (16).
The present study aimed to examine the potential
relevance of various mechanisms underlying the low levels of RKIP
exhibited by the SUM 159 TNBC cell line.
Materials and methods
Reagents
The demethylating agent 5-aza-2′-deoxycytidine
(5-AZA) and the histone deacetylase (HDAC) inhibitor trichostatin A
(TSA) were purchased from Sigma-Aldrich Srl, Milan, Italy. The
NF-κB inhibitor dehydroxymethylepoxyquinomicin (DHMEQ) was provided
by Professor Kazuo Umezawa (Department of Applied Chemistry,
Faculty of Science and Technology, Keio University, Yokohama,
Japan).
Cell cultures
The BT 549, MCF 7, MCF 7R and MDA MB 231 cell lines
were cultured in RPMI-1640, the MDA MB 468 cell line was cultured
in Dulbecco's modified Eagle's medium (DMEM), the SUM 159 and SUM
149 cell lines were cultured in DMEM/F-12 supplemented with insulin
(5 µg/ml), and the MCF 10A cell line was cultured in DMEM/F-12
supplemented with insulin (0.01 mg/ml), epidermal growth factor (20
ng/ml) and hydrocortisone (500 ng/ml). All media were supplemented
with 10% heat-inactivated fetal calf serum, 2 mM L-glutamine, 100
U/ml penicillin and 100 µg/ml streptomycin (all reagents were from
EuroClone S.p.A., Milan, Italy; GE Healthcare Life Sciences, Logan,
UT, USA). The cells were cultured in a humidified atmosphere at
37°C in 5% CO2. After obtaining the cells, the first
passage carried out was assigned passage number 1. Cells with a
narrow range of passage number (4–6) that were
routinely tested for Mycoplasma contamination were used for all
experiments. The breast carcinoma MCF-7 line was purchased from the
American Type Culture Collection (Manassas, VA, USA) and its
multi-drug resistant variant MCF-7R was established by treating the
wild-type MCF-7 cells with gradually increasing concentrations of
doxorubicin. Non-malignant MCF 10A breast epithelial cells were
provided by Dr. Agata Giallongo (Institute of Biomedicine and
Molecular Immunology, National Research Council, Palermo, Italy)
and TNBC cell lines were provided by Dr. Elda Tagliabue (Molecular
Targeting Unit, Department of Experimental Oncology and Molecular
Medicine, Fondazione Institute of Hospitalization and Scientific
Care, National Cancer Institute, Milan, Italy).
Cell growth assays
The cells were seeded at 2×104 cells/well
onto 96-well plates and incubated overnight at 37°C. At time 0, the
medium was replaced with fresh complete medium supplemented with
5-AZA, TSA, DHMEQ or combinations of AZA + TSA or DHMEQ + TSA at
the indicated concentrations. Following 72 h of treatment, 15 µl
commercial solution obtained from Promega Corp. (Madison, WI, USA)
containing
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium
(MTS) and phenazine ethosulfate was added. The plates were
incubated in a humidified atmosphere at 37°C in 5% CO2
for 2 h, and the bioreduction of MTS dye was evaluated by measuring
the absorbance of each well at 490 nm using a microplate absorbance
reader (iMark Microplate Reader; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Cell growth inhibition was expressed as a
percentage of the absorbance of the control cells. For the
combinations, to evaluate the type of interaction between the
agents, dose-effect curves were analyzed according to the Chou and
Talalay method (17) using CalcuSyn®
software (version 2.1; Biosoft, Cambridge, UK) as a non-constant
ratio combination. The combination index (CI) indicates a
quantitative measure of the degree of drug interaction in terms of
synergistic (CI<1), additive (CI=1) or antagonistic (CI>1)
effects.
Evaluation of cell death by flow
cytometry
SUM 159 cells were treated with the agents alone or
in combination for 48 h. Subsequently, the cells were washed twice
with ice-cold phosphate-buffered saline (PBS; EuroClone S.p.A.) and
then suspended at a density of 1×106 cells/ml in a
hypotonic fluorochrome solution containing 50 µg/ml propidium
iodide in 0.1% sodium citrate with 0.03% (v/v) Nonidet P-40
(Sigma-Aldrich Srl). Following incubation in this solution for 1 h,
the samples were filtered through a 40-µm mesh nylon cloth, and
fluorescence was evaluated as single-parameter frequency histograms
using a FACSort instrument (BD Biosciences, San Jose, CA, USA). The
data were analyzed using CellQuest™ software (version 3.1; BD
Biosciences). Cell death was measured by determination of the
percentage of events accumulated in the pre-G0-G1 position.
DNA extraction and bisulphite
modification
Ten breast cancer samples were obtained from the
Anatomic Pathology Unit, University Hospital ‘Paolo Giaccone’
(Palermo, Italy) between 2013 and 2014. This study was approved by
the ethics commitee of the University of Palermo (Palermo, Italy)
and written informed consent was obtained from all patients. Two
8-µm sections microdissected from each of the ten paraffin-embedded
samples. Cell lysis and DNA extraction were performed on the ten
samples, as well as SUM 159 cells, using a QIAamp DNA mini kit
(Qiagen GmbH, Hilden Germany) according to the manufacturer's
protocol. Extracted genomic DNA was diluted in 50 µl distilled
water. Sodium bisulphite conversion was performed using the EpiTect
Plus Bisulfite Conversion kit (Qiagen GmbH) using 500 ng DNA,
according to the manufacturer's protocol.
Immunohistochemical analysis
Evaluation of the positivity for Ki-67, ER and PR,
HER2 and RKIP protein was performed by immunohistochemistry, as
previously described (18,19). Briefly, tissue samples were fixed in
10% buffered formalin and paraffin-embedded. Tissue sections (4 µm)
were then deparaffinized and rehydrated. The sections were
pretreated with Tris/EDTA buffer (pH 9.0; Novocastra Reagents,
Milton Keynes, UK) and Cell Condition Solution (Ventana Medical
Systems, Inc., Tucson, AZ, USA) in a PT Link pre-treatment system
(Dako UK Ltd., Ely, UK) at 98°C for 30 min. Subsequently, the
sections were washed in PBS at room temperature. The endogenous
peroxidase was neutralized using 3% H2O2 and
the membranes were blocked using 4% casein (Novocastra Reagents).
The samples were then incubated for 1 h at room temperature with
the following primary antibodies: Polyclonal rabbit anti-human RKIP
(1:100; cat. no 4742; Cell Signaling Technology Inc., Danvers, MA,
USA), monoclonal rabbit anti-human estrogen receptor, (clone SP1;
1:100; cat. no. 790–4324; Ventana Medical Systems), monoclonal
rabbit anti-human progesterone receptor (clone 1E2; 1:100; cat. no.
790–2223; Ventana Medical Systems), rabbit anti-human monoclonal
Ki-67 (clone 30-9; 1:500; cat. no. 790–4286; Ventana Medical
Systems). Staining was detected using a polymer detection kit
(Novocastra Reagents) and 3,3′-diaminobenzidine substrate-chromogen
(Novocastra Reagents) and the slides were counterstained using
Harris hematoxylin (Novocastra Reagents).
Methylation-specific polymerase chain
reaction (PCR)
Methylation of the RKIP gene promoter was
investigated using methylation-specific PCR. The reaction was
performed in a total volume of 50 µl, comprised of 6 µl bisulphite
modified DNA, 0.2 µM each sense and anti-sense primers (Invitrogen
Life Technologies, Carlsbad, CA, USA), 1 µl dNTPs (0.2 mM each),
1,5 mM MgCl2, 1X PCR buffer and 1 unit Platinum Taq DNA
Polymerase (Invitrogen Life Technologies). The cycling conditions
were as follows: Initial denaturation at 95°C for 10 min, followed
by 40 cycles of denaturation at 95°C for 1 min, annealing at 52°C
for 1 min and extension at 72°C for 1 min, followed by a final
extension step at 72°C for 10 min. To differentiate between the
methylated DNA (204 bp PCR product) and the unmethylated DNA (205
bp PCR product), the following specific primers were used:
Unmethylated RKIP promoter forward, 5′-TTTAGTGATATTTTTTGAGATATGA-3′
and reverse, 3′-CACTCCCTAACCTCTAATTAACCAA-5′; methylated RKIP
promoter forward, 5′-TTTAGCGAT ATTTTTTGAGATACGA-3′ and reverse,
3′-GCTCCCTAA CCTCTAATTAACCG-5′ (Applied Biosystems Life
Technologies, Foster City, CA, USA).
CpGenome universal methylated DNA (Chemicon
International, Inc., Temecula, CA, USA) was used as a methylated
positive control. Blood DNA obtained from a healthy, young
individual was used as an unmethylated negative control following
receipt of written informed consent.
Cell transfection with anti-miR
microRNA (miRNA) inhibitor and pre-miR miRNA precursor
SUM 159 cells were transfected with 30 nM
pre-miR-224 miRNA precursor, anti-miR-224 miRNA inhibitor and
relative random sequences (pre-miR negative control) as negative
controls, using siPORT™ Amine Transfection Agent (all these
reagents were from Ambion Life Technologies, Carlsbad, CA, USA),
according to the manufacturer's instructions.
Extraction of cellular RNA and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from clinical samples and
cell lines using TRIzol reagent (Invitrogen Life Technologies). For
the evaluation of miR-224 levels, mature miRNA was reverse
transcribed using miRNA-specific stem-loop primers (TaqMan MicroRNA
Reverse Transcription kit; Applied Biosystems Life Technologies)
prior to qPCR, which was conducted according to the manufacturer's
instructions (TaqMan MicroRNA assay; Applied Biosystems Life
Technologies) on a StepOne RealTime PCR System (Applied Biosystems
Life Technologies). The PCR cycling conditions were as follows:
Denaturation at 50°C for 2 min, annealing at 95°C for 10 min,
followed by 40 cycles of 95°C for 15 sec and extension at 60°C for
60 min. Samples were normalized to RNU6B small RNA.
For the evaluation of RKIP expression, RNA was
reverse transcribed using a high capacity complementary DNA (cDNA)
reverse transcription kit (Applied Biosystems Life Technologies).
The resulting cDNAs were subjected to real-time RT-PCR using the
TaqMan Gene Expression Master Mix kit (Applied Biosystems Life
Technologies) in triplicates. The PCR cycling conditions were as
follows: Denaturation at 50°C for 2 min, annealing at 95°C for 10
min, followed by 40 cycles of 95°C for 15 sec and extension at 60°C
for 60 min. The running of the samples and data collection were
performed on a StepOne AB Real Time PCR system (Applied Biosystems
Life Technologies). β-actin was used as an internal standard. The
specific primers used were as follows: hsa-miR-224, 002099; RNU 6B,
001093; RKIP, Hs01110783_g1; and β-actin, Hs99999903_m1 (Applied
Biosystems Life Technologies).
Relative expression was calculated using the
comparative Ct method [ΔCt=Ct(target
gene)-Ct(housekeeping gene)]. Where Ct was the
fractional cycle number at which the fluorescence of each sample
passed the fixed threshold. Fluorescence was measured at 515–518 nm
using StepOne AB Real Time PCR System software (Applied Biosystems
Life Technologies). The ΔΔCt method for relative quantification of
gene expression was used to determine miRNA or gene expression
levels. ΔΔCt was calculated using the formula: ΔΔCt=ΔCt(each
sample)-ΔCt(reference sample). Fold change was
calculated using the 2−ΔΔCt equation.
Western blotting
Whole-cell lysates were obtained from breast cancer
cells using radioimmunoprecipitation assay buffer (Santa Cruz
Biotechnology Inc., Dallas, TX, USA) and 25 µg protein was
subjected to 10% SDS-PAGE and transferred to Hybond-P membranes (GE
Healthcare Europe GmbH, Freiburg, Germany). Filters were incubated
with primary antibodies raised against β-actin (1:10,000;
Sigma-Aldrich Srl) or RKIP (1:1,000; Cell Signaling Technology,
Inc.). Immunoblots were quantified by densitometry and results were
expressed as arbitrary units (RKIP/β-actin).
Cell invasion assay
Cell invasion assays were performed using BD BioCoat
Matrigel Invasion Chambers (BD Biosciences). SUM 159 cells were
seeded at a density of 4×105 cells/well onto 6-well
plates. Following 24 h of incubation, 5-AZA (1.5 mM) or TSA (1 µM)
was added. Following 16 h of treatment, the cells were trypsinized
(EuroClone S.p.A.) and transferred to the upper Matrigel chamber in
500 µl medium at a density of 2.5×104. Medium
supplemented with 10% fetal bovine serum (EuroClone S.p.A.) was
added to the lower chamber. Following 48 h of incubation, the
non-migrated cells in the upper chamber were carefully removed with
a cotton tip and the adherent cells present on the lower surface of
the Matrigel insert were fixed with 100% methanol and stained with
Giemsa solution (1:10; Sigma-Aldrich Srl). Migrated cells were
counted microscopically (CK2 microscope; Olympus, Tokyo, Japan) in
four randomly selected fields of the membrane at a magnification of
×40. Each invasion experiment was carried out in duplicate and
repeated at least twice. Data are expressed as a percentage of
invasion through the Matrigel matrix, relative to that of the
untreated controls. In parallel, cell growth assays were performed
under identical conditions.
NF-κB activation
The DNA-binding capacity of NF-κB (p65 subunit) was
determined in the nuclear extracts of SUM 159 cells using the
TransAM™ NF-κB and Nuclear Extract™ kits (Active Motif, Carlsbad,
CA, USA) according to the manufacturer's instructions. The SUM 159
cells were treated with 10 or 20 µg/ml DHMEQ for 8 or 16 h.
Briefly, the determination of binding capacity was based on a
96-well plate, upon which an oligonucleotide containing the NF-κB
consensus binding site (5′-GGGACTTTCC-3′) was immobilized.
Activated NF-κB contained in the extracts is able to specifically
bind to this nucleotide. NF-κB bound to the oligonucleotide may
subsequently be detected using an antibody directed against an
epitope on p65 (polyclonal rabbit anti-human; cat. no. 40096;
1:1,000; Active Motif), accessible only when NF-κB is bound to its
target DNA.
Subsequently, the addition of a horseradish
peroxidase-conjugated secondary antibody provided a sensitive
colorimetric readout that may be quantified by densitometry (iMark
Microplate Reader; Bio-Rad Laboratories, Inc.). The specificity of
the assay was confirmed by simultaneous incubations in the presence
of excess, non-immobilized consensus oligonucleotides, as a
competitor, or of a mutated consensus oligonucleotide. The results
were expressed as arbitrary units: one unit indicated the DNA
binding capacity exerted by 2.5 µg whole cell extract from Jurkat
cells (positive control for NF-κB p65 activation; Active Motif)
(stimulated with 12-O-tetradecanoylphorbol-13-acetate and
calcium ionophore) per microgram of protein from the nuclear
extracts.
Statistical analysis
Results are presented as the mean ± standard
deviation. The significance of differences between means was
evaluated by Student's t-test for unpaired samples. The association
between miR-224 and RKIP mRNA or protein levels (Table I) was evaluated by calculating
Pearson's correlation coefficient (r). P<0.05 indicated a
statistically significantly difference.
 | Table I.RKIP and miR-224 expression levels in
various breast cancer cell lines. |
Table I.
RKIP and miR-224 expression levels in
various breast cancer cell lines.
| Cell line | RKIP protein
levels | RKIP mRNA
levels | miR-224 levels |
|---|
| MDA 231 | 0.717 | 0.500 | 0.815 |
| MDA 468 | 0.980 | 0.768 | 0.013 |
| BT 549 | 0.686 | 0.136 | 0.101 |
| SUM 149 | 1.131 | 0.866 | 3.964 |
| SUM 159 | 0.559 | 0.402 | 0.923 |
| MCF 10A | 1.063 | 1.000 | 1.000 |
| MCF 7 | 1.667 | 1.130 | 0.001 |
| MCF 7R | 1.069 | 0.390 | 0.000 |
Results
RKIP expression levels are low in the
SUM 159 breast cancer cell line
The RKIP content of the non-malignant MCF 10A breast
epithelial cells and of several breast cancer cell lines, including
MCF 7 and its multi-drug resistant variant MCF 7R, as well as the
TNBC MDA MB 231, MDA MB 468, BT 549, SUM 149, SUM 159 and MCF 10A
cell lines were evaluated (Table I).
Together with BT 549, the SUM 159 cell line was found to express
low levels of RKIP, at the mRNA and protein level, and therefore
this cell line was selected for further analysis of the potential
mechanisms involved in the repression of RKIP. SUM 159 cells are
highly invasive, originate from a primary anaplastic, grade 4
carcinoma and belong to the basal B mesenchymal stem-like subtype
according to the classification outlined by Neve et al
(20).
Demethylating agent 5-AZA demethylates
the RKIP gene promoter, increases RKIP expression and inhibits
invasion of SUM 159 cells
It has been debated whether epigenetic changes,
including methylation of the RKIP gene promoter, may be responsible
for RKIP downregulation in colorectal and other types of cancer
(11,21–24).
Methylation-specific PCR was performed on 10 primary
ductal infiltrating breast cancers of various subtypes and with
differing grades of RKIP expression (Table II) and in SUM 159 cells (Fig. 1). Gene promoters were considered
methylated if the intensity of methylated bands was >10% of
their respective unmethylated bands (25). Five of the clinical tumors exhibited
RKIP gene promoter hypermethylation, which was associated with the
low expression of RKIP protein and mRNA levels. In addition, SUM
159 cells demonstrated hypermethylation of the RKIP promoter
(Fig. 1). The SUM 159 cells were
subsequently exposed to various concentrations (500 µM, 1 and 2 mM)
of demethylating agent 5-AZA for 72 h. The 5-AZA reagent was able
to demethylate the RKIP gene promoter in a dose-dependent manner
(Fig. 1) and to increase RKIP
expression at the mRNA and protein level (Table III). Conversely, in cell invasion
assays, 5-AZA, at a concentration of 1.5 mM, which was only
marginally cytotoxic (3.7±0.2% inhibition of cell growth), markedly
inhibited (82.5±2.1%) the invasion of SUM 159 cells.
| Table II.Characteristics of ten breast cancer
samples. |
Table II.
Characteristics of ten breast cancer
samples.
| Sample | Histotype | Grading | Ki-67 | ER | PR | HER2 | Methylation of
RKIP gene promoter | RKIP mRNA
levels | Lymph RKIP
protein | node
metastasis |
|---|
| T1 | Luminal A | G1 | Pos<5% | + | + | − | + | 0.058 | − | − |
| T2 | Luminal A | G2 | Pos<10% | + | + | − | + | 0.279 | − | − |
| T3 | Luminal A | G2 | Pos<10% | + | + | − | + | 0.352 | − | + |
| T4 | Luminal B | G2 | Pos<10% | + | + | + | + | 0.426 | − | − |
| T5 | Luminal A | G2 | Pos<10% | + | + | − | + | 0.355 | − | − |
| T6 | TN | G3 | Pos<10% | − | − | − | − | 1.068 | + | + |
| T7 | Luminal B | G3 | Pos>10% | + | + | + | − | 0.477 | + | − |
| T8 | TN | G3 | Pos>10% | − | − | − | − | 0.493 | + | − |
| T9 | TN | G3 | Pos>10% | − | − | − | − | 0.627 | + | + |
| T10 | Basal-like CK
5/6+ | G3 | Pos>50% | − | − | − | − | 0.494 | + | + |
| Table III.RKIP mRNA and protein levels in SUM
159 cells following treatment with 5-AZA, TSA or a combination of
the two for 72 h. |
Table III.
RKIP mRNA and protein levels in SUM
159 cells following treatment with 5-AZA, TSA or a combination of
the two for 72 h.
| Conditions | RKIP mRNA | RKIP protein |
|---|
| Control | 1.000±0.000 | 0.642±0.093 |
| 5-AZA |
1.454±0.111b |
1.010±0.107b |
| TSA |
1.476±0.206a |
1.184±0.085b |
| 5-AZA+TSA |
1.507±0.099b |
1.011±0.145a |
HDAC inhibitor TSA enhances RKIP
expression and inhibits invasion in SUM 159 cells
It is well known that another epigenetic mechanism
that affects gene expression is histone acetylation (26). Notably, the HDAC inhibitor TSA was
able to increase the mRNA and protein expression levels of RKIP
(Table III) in SUM 159 cells, as
well as exert inhibitory effects in the specific assays determining
their invasion ability (45.9±3.1% at 1 µM, which induced 12.2±1.6%
cell growth inhibition).
The combination of 5-AZA and TSA analyzed was not
able to further increase the effects of either agent on RKIP mRNA
and protein expression levels (Table
III). The effects of 5-AZA and TSA alone or in combination on
the inhibition of cell growth were evaluated using MTS assays
(Tables IV and V). 5-AZA and TSA treatment both inhibited
cell growth in a dose dependent manner (Table IV). The combinatory effects were
found to be substantially additive, as indicated by the combination
indices (Table V). Furthermore, flow
cytometric analysis of the induction of apoptosis revealed that,
overall, the combination of 5-AZA+TSA produced only modest
increases in apoptosis with respect to the expected sum, based on
the effects of the agents alone (Table
VIA).
| Table IV.Results of cell growth assays of SUM
159 cells following treatment with various concentrations of 5-AZA,
TSA or DHMEQ. |
Table IV.
Results of cell growth assays of SUM
159 cells following treatment with various concentrations of 5-AZA,
TSA or DHMEQ.
| Treatment | Cell growth
inhibition, % |
|---|
| 5-AZA, mM |
|
|
0.5 |
1.1±0.2b |
|
0.75 |
6.0±2.0b |
|
1.0 |
5.2±1.9a |
|
2.0 |
40.1±4.9b |
| TSA, µM |
|
|
0.5 |
5.8±2.9a |
|
0.75 |
19.2±3.7b |
|
1.0 | 24.
5±4.9b |
|
1.5 |
43.1±4.4b |
|
2.0 |
83.1±2.1b |
| DHMEQ, µg/ml |
|
|
5.0 | 0.0 |
|
7.5 | 0.2±0.2 |
| 10 | 0.3±0.5 |
| 15 | 2.5±2.0 |
| 20 |
2.0±0.7b |
| Table V.Cell growth inhibition and
combination indices in SUM 159 cells following combination
treatment with various concentrations of 5-AZA, TSA and DHMEQ. |
Table V.
Cell growth inhibition and
combination indices in SUM 159 cells following combination
treatment with various concentrations of 5-AZA, TSA and DHMEQ.
| Treatment | Cell growth
inhibition, % | Combination
index |
|---|
| 5-AZA (0.5 mM)+TSA
(0.5 µM) |
20.2±2.4a | 0.941 |
| 5-AZA (0.5 mM)+TSA
(1.0 µM) |
34.9±4.1a | 1.175 |
| 5-AZA (0.5 mM)+TSA
(1.5 µM) |
61.9±5.8a | 1.105 |
| 5-AZA (0.75 mM)+TSA
(0.5 µM) |
21.3±3.2a | 1.089 |
| 5-AZA (0.75 mM)+TSA
(1.0 µM) |
34.3±2.6a | 1.327 |
| 5-AZA (0.75 mM)+TSA
(1.5 µM) |
69.8±5.3a | 1.053 |
| 5-AZA (1.0 mM)+TSA
(0.5 µM) |
17.1±3.0a | 1.376 |
| 5-AZA (1.0 mM)+TSA
(1.0 µM) |
47.3±2.3a | 1.206 |
| 5-AZA (1.0 mM)+TSA
(1.5 µM) |
80.0±3.7a | 0.936 |
| 5-AZA (2.0 mM)+TSA
(0.5 µM) |
54.8±6.0a | 1.129 |
| 5-AZA (2.0 mM)+TSA
(1.0 µM) |
87.2±3.3a | 0.810 |
| 5-AZA (2.0 mM)+TSA
(1.5 µM) |
99.3±0.7a | 0.342 |
| DHMEQ (7.5
µg/ml)+TSA (0.5 µM) |
35.1±2.9a | 0.662 |
| DHMEQ (7.5
µg/ml)+TSA (1.5 µM) |
64.9±6.7a | 0.716 |
| DHMEQ (10
µg/ml)+TSA (0.5 µM) |
41.7±6.5a | 0.659 |
| DHMEQ (10
µg/ml)+TSA (1.0 µM) |
77.2±5.7a | 0.608 |
| DHMEQ (15
µg/ml)+TSA (0.5 µM) |
72.0±2.7a | 0.482 |
| DHMEQ (15
µg/ml)+TSA (1.0 µM) |
87.9±2.3a | 0.507 |
| DHMEQ (20
µg/ml)+TSA (0.5 µM) |
88.5±1.5a | 0.366 |
| DHMEQ (20
µg/ml)+TSA (1.0 µM) |
84.2±3.1a | 0.628 |
| Table VI.Flow cytometric analysis of apoptosis
in SUM 159 cells following treatment with 5-AZA, TSA, DHMEQ or a
combination of these. |
Table VI.
Flow cytometric analysis of apoptosis
in SUM 159 cells following treatment with 5-AZA, TSA, DHMEQ or a
combination of these.
| A, 5-AZA and
TSA |
|
|
|---|
|
|---|
| Conditions | Apoptosis, % | Expected, % |
|---|
| Control | 3.7±0.1 |
|
| 5-AZA, mM |
|
|
|
2.0 | 16.9±1.3 |
|
|
3.0 | 41.2±2.0 |
|
| TSA, µM |
|
|
|
1.0 | 18.3±0.6 |
|
|
2.0 | 41.2±1.8 |
|
| Combinations |
|
|
| 5-AZA
(2.0 mM)+TSA (1.0 µM) | 37.2±2.5 | 31.5±0.6 |
| 5-AZA
(2.0 mM)+(TSA 2.0 µM) | 79.8±5.4 |
54.4±3.0a |
| 5-AZA
(3.0 mM)+TSA (1.0 µM) | 64.5±1.6 | 56.0±2.7 |
| 5-AZA
(3.0 mM)+TSA (2.0 µM) | 81.3±2.8 | 78.9±0.3 |
|
| B, DHMEQ and
TSA |
|
|
|
| Conditions | Apoptosis, % | Expected, % |
|
| Control | 3.8±0.1 |
|
| DHMEQ, µg/ml |
|
|
| 15 | 15.2±1.1 |
|
| 20 | 18.5±0.1 |
|
| TSA, µM |
|
|
|
1.0 | 19.8±1.2 |
|
|
1.5 | 25.4±0.5 |
|
| Combinations |
|
|
| DHMEQ (15
µg/ml)+TSA (1.0 µM) | 59.4±1.9 |
31.1±2.4b |
| DHMEQ (15
µg/ml)+TSA (1.5 µM) | 62.2±1.7 |
36.8±1.8b |
| DHMEQ (20
µg/ml)+TSA (1.0 µM) | 64.3±0.8 |
34.5±1.2b |
| DHMEQ (20
µg/ml)+TSA (1.5 µM) | 74.6±2.4 |
40.1±0.5b |
DHMEQ reduces constitutive activation
of NF-κB and increases RKIP levels in SUM 159 cells
Previous studies have indicated that activated NF-κB
is able to downregulate RKIP expression via Snail induction
(13,14,27).
Evaluation by TransAM® assays revealed that SUM 159 cells exhibited
a marked constitutive activation of NF-κB, which was reduced in a
concentration-dependent manner by DHMEQ, an inhibitor of the
nuclear translocation of this transcription factor (Fig. 2) (28,29).
Furthermore, identical treatments with DHMEQ induced marked
increases in RKIP mRNA and protein expression in SUM 159 cells
(Table VII).
| Table VII.RKIP mRNA and protein levels of SUM
159 cells following treatment with DHMEQ for 8 or 16 h. |
Table VII.
RKIP mRNA and protein levels of SUM
159 cells following treatment with DHMEQ for 8 or 16 h.
| Conditions | Duration, h | RKIP mRNA | RKIP protein |
|---|
| Control | 0 | 1.000 | 0.654 |
| DHMEQ, µg/ml |
|
|
|
| 10 | 8 | 1.218 | 0.983 |
| 20 | 8 | 1.302 | 0.870 |
| 10 | 16 | 1.375 | 1.780 |
| 20 | 16 | 1.285 | 1.537 |
Notably, combined treatment with DHMEQ and TSA
produced synergistic effects on the inhibition of cell growth
(Table V) and induction of apoptosis
(Table VIB).
miR-224 does not alter RKIP expression
levels
Upregulation of miRNAs has previously been
associated with cancer progression, via the inhibition of tumor
suppressor genes (30,31). Furthermore, a previous study (16) demonstrated that miR-224 was able to
inhibit RKIP gene expression by directly targeting the
3′-untranslated region of the highly invasive MDA MB 231 breast
cancer cell line.
However, by calculating the Pearson's correlation
coefficient, a significant inverse association between miR-224 and
RKIP levels could not be found in the breast cancer cell lines
evaluated in the present study (Table
I).
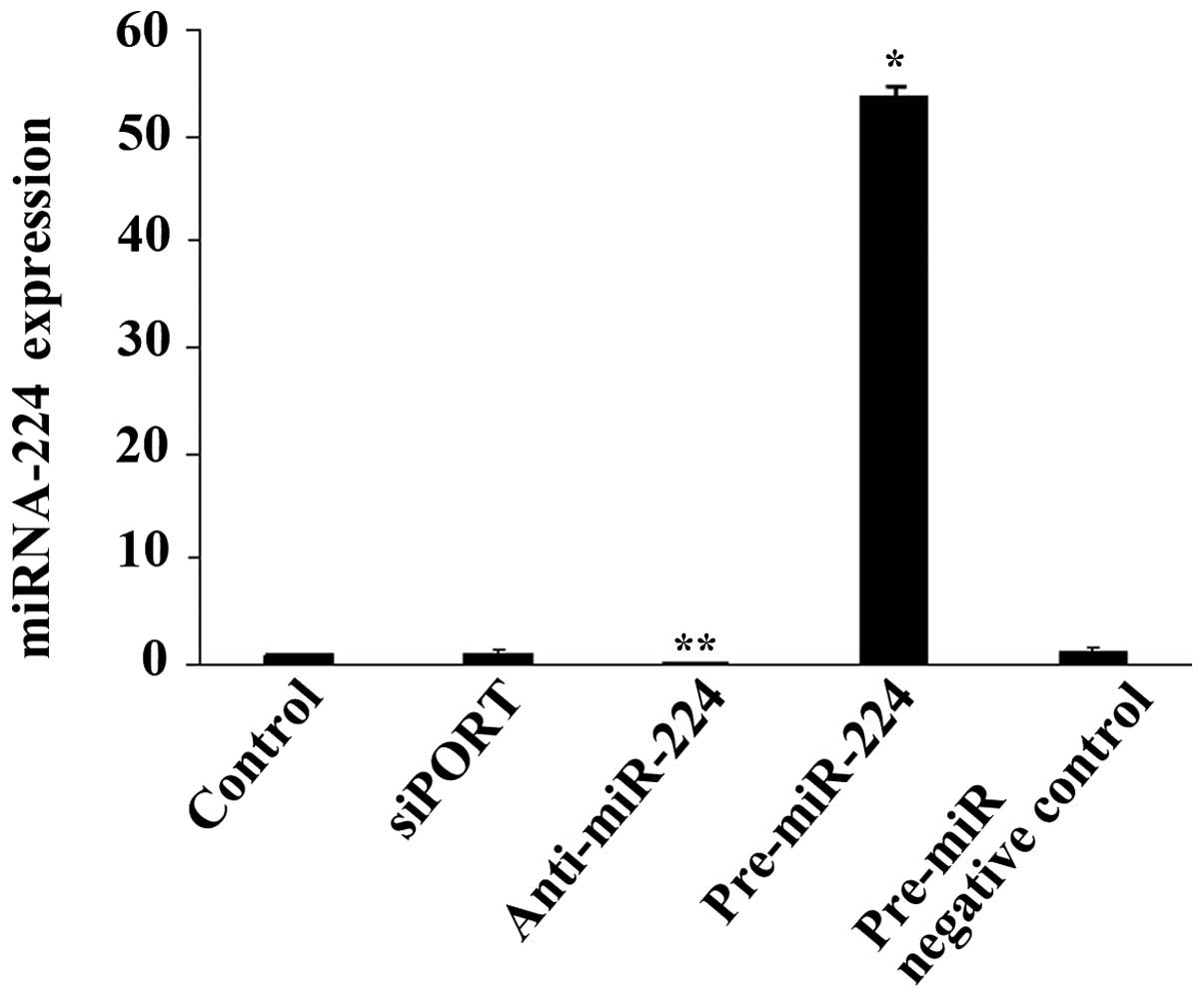
Furthermore, SUM 159 cells were transfected with
pre-miR-224 miRNA precursor, anti-miR-224 miRNA inhibitor or siPORT
Amine Transfection Agent alone or with a negative control for 24 h.
The increase or reduction in miR-224 (Fig. 3) induced by the precursor or
inhibitor, respectively, did not alter RKIP expression at the mRNA
or protein level (data not shown).
Discussion
Overall, the present results suggest that various
mechanisms, including methylation of the gene promoter, histone
deacetylation and NF-κB activation, but not targeting by miR-224,
may be responsible for the downregulation of RKIP gene expression
in the highly invasive SUM 159 TNBC cell line. However, previous
studies of hepatocellular carcinoma cell lines (11,32) ruled
out the role of gene methylation, histone acetylation and miR-224
in the reduction of RKIP expression. These results highlight the
complexity of the regulation of this tumor suppressor gene, which
may be differentially affected depending on the type of cancer.
From a therapeutic perspective, the demethylating agent 5-AZA, the
HDAC inhibitor TSA and the NF-κB inhibitor DHMEQ, in addition to
inducing an increase in RKIP expression, appeared to be able to
generate significant cytotoxicity, induce apoptosis and, in the
case of 5-AZA and TSA, reduce cell invasion. Results in other cell
models have demonstrated that the combination of DNA demethylating
agents and HDAC inhibitors, for example 5-AZA and TSA, may be
synergistic in the reactivation of specific genes (33,34).
However, this did not appear to be the case for RKIP in SUM 159
cells. Furthermore, 5-AZA and TSA generated mainly additive effects
on cell growth inhibition and induction of apoptosis when the cells
were treated with the two agents together. The combination of DHMEQ
and TSA exhibited significant synergy in cell growth and induction
of apoptosis assays. This was unsurprising, since, besides
influencing RKIP expression, 5-AZA, TSA and DHMEQ may distinctly
influence additional genes, and therefore their respective
properties may determine their respective capabilities of
synergizing with other agents, with regard to the inhibition of
cell growth, survival and invasive and metastatic ability.
Concurrently, it was previously demonstrated that TSA and DHMEQ
were able to diversely alter gene expression and synergize with
conventional chemotherapeutic drugs, including doxorubicin and
cisplatin, in hepatocellular cancer cells (29,32).
The present results, in addition to confirming the
potential significance of NF-κB activation in the downregulation of
RKIP expression (13,14,27),
suggest that this transcription factor may be a useful target for
the treatment and chemosensitization of the most aggressive forms
of breast cancer. There is increasing evidence that aberrant
activation of NF-κB signaling is a frequent characteristic of TNBC
cells, although the underlying causes of this activation have
remained largely elusive (35,36). Apart
from DHMEQ, which at preclinical levels appears to be a promising
agent for anticancer, anti-inflammatory and immunosuppressive
treatments (28), other valuable
therapeutic resources in this context may include the proteasome
inhibitors, which, in addition to inhibiting NF-κB activation, may
also restore RKIP levels via inhibition of proteasome degradation
of the ubiquinated protein (11,37).
In conclusion, the present findings, in addition to
other potential causes of RKIP downregulation, require further
investigation and validation in a larger number of TNBC cell
models.
Acknowledgements
The present study was supported by the Fund for
Investment in Basic Research (FIRB) of Medical Research in Italy
(MERIT) (no. RBNE08YYBM_004) and the Finalized Research Fund (FFR;
no. 2012/2013 2012-ATE-0509).
References
|
1
|
Schmadeka R, Harmon BE and Singh M:
Triple-negative breast carcinoma: Current and emerging concepts. Am
J Clin Pathol. 141:462–477. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abramson VG, Lehmann BD, Ballinger TJ and
Pietenpol JA: Subtyping of triple-negative breast cancer:
Implications for therapy. Cancer. 121:8–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Minn AJ, Bevilacqua E, Yun J and Rosner
MR: Identification of novel metastasis suppressor signaling
pathways for breast cancer. Cell Cycle. 11:2452–2457. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hao C, Wei S, Tong Z, Li S, Shi Y, Wang X
and Zhu ZH: The effects of RKIP gene expression on the biological
characteristics of human triple-negative breast cancer cells in
vitro. Tumour Biol. 33:1159–1167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Al-Mulla F, Bitar MS, Thiery JP, Zea TT,
Chatterjee D, Bennett L, Park S, Edwards J and Yeung KC: Clinical
implications for loss or diminution of Raf-1 kinase inhibitory
protein and its phosphorylated form in ductal breast cancer. Am J
Cancer Res. 3:446–464. 2013.PubMed/NCBI
|
|
6
|
Odabaei G, Chatterjee D, Jazirehi AR,
Goodglick L, Yeung K and Bonavida B: Raf-1 kinase inhibitor
protein: Structure, function, regulation of cell signaling and
pivotal role in apoptosis. Adv Cancer Res. 91:169–200. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang H, Park S, Sun SC, Trumbly R, Ren G,
Tsung E and Yeung KC: RKIP inhibits NF-kappaB in cancer cells by
regulating upstream signaling components of the IkappaB kinase
complex. FEBS Lett. 584:662–668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yousuf S, Duan M, Moen EL, Cross-Knorr S,
Brilliant K, Bonavida B, LaValle T, Yeung KC, Al-Mulla F, Chin E
and Chatterjee D: Raf kinase inhibitor protein (RKIP) blocks signal
transducer and activator of transcription 3 (STAT3) activation in
breast and prostate cancer. PLoS One. 9:e924782014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eves EM, Shapiro P, Naik K, Klein UR,
Trakul N and Rosner MR: Raf kinase inhibitory protein regulates
aurora B kinase and the spindle checkpoint. Mol Cell. 23:561–574.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klysik J, Theroux SJ, Sedivy JM, Moffit JS
and Boekelheide K: Signaling crossroads: The function of Raf kinase
inhibitory protein in cancer, the central nervous system and
reproduction. Cell Signal. 20:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Poma P, Labbozzetta M, Vivona N, Porcasi
R, D'Alessandro N and Notarbartolo M: Analysis of possible
mechanisms accounting for raf-1 kinase inhibitor protein
downregulation in hepatocellular carcinoma. OMICS. 16:579–588.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jazirehi AR and Bonavida B: Cellular and
molecular signal transduction pathways modulated by rituximab
(rituxan, anti-CD20 mAb) in non-Hodgkin's lymphoma: Implications in
chemosensitization and therapeutic intervention. Oncogene.
24:2121–2143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hsu YL, Chen CY, Lin IP, Tsai EM, Kuo PL
and Hou MF: 4-Shogaol, an active constituent of dietary ginger,
inhibits metastasis of MDA-MB-231 human breast adenocarcinoma cells
by decreasing the repression of NF-κB/Snail on RKIP. J Agric Food
Chem. 60:852–861. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen TC, Hsu YL, Tsai YC, Chang YW, Kuo PL
and Chen YH: Gemifloxacin inhibits migration and invasion and
induces mesenchymal-epithelial transition in human breast
adenocarcinoma cells. J Mol Med (Berl). 92:53–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ren G, Baritaki S, Marathe H, Feng J, Park
S, Beach S, Bazeley PS, Beshir AB, Fenteany G, Mehra R, et al:
Polycomb protein EZH2 regulates tumor invasion via the
transcriptional repression of the metastasis suppressor RKIP in
breast and prostate cancer. Cancer Res. 72:3091–3104. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang L, Dai T, Lin X, Zhao X, Chen X,
Wang C, Li X, Shen H and Wang X: MicroRNA-224 targets RKIP to
control cell invasion and expression of metastasis genes in human
breast cancer cells. Biochem Biophys Res Commun. 425:127–133. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chou TC and Talalay P: Analysis of
combined drug effects: A new look at a very old problem. Trends
Pharmacol Sci. 4:450–454. 1983. View Article : Google Scholar
|
|
18
|
Florena AM, Tripodo C, Guarnotta C, Ingrao
S, Porcasi R, Martorana A, Lo Bosco G, Cabibi D and Franco V:
Associations between Notch-2, Akt-1 and HER2/neu expression in
invasive human breast cancer: A tissue microarray immunophenotypic
analysis on 98 patients. Pathobiology. 74:317–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Notarbartolo M, Giannitrapani L, Vivona N,
Poma P, Labbozzetta M, Florena AM, Porcasi R, Muggeo VM, Sandonato
L and Cervello M: Frequent alteration of the Yin Yang 1/Raf-1
kinase inhibitory protein ratio in hepatocellular carcinoma. OMICS.
15:267–272. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Neve RM, Chin K, Fridlyand J, Baehner FL,
Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, et al: A
collection of breast cancer cell lines for the study of
functionally distinct cancer subtypes. Cancer Cell. 10:515–527.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schuierer MM, Bataille F, Weiss TS,
Hellerbrand C and Bosserhof AK: Raf kinase inhibitor protein is
down-regulated in hepatocellular carcinoma. Oncol Rep. 16:451–456.
2006.PubMed/NCBI
|
|
22
|
Minoo P, Zlobec I, Baker K, Tornillo L,
Terracciano L, Jass JR and Lugli A: Loss of raf-1 kinase inhibitor
protein expression is associated with tumor progression and
metastasis in colorectal cancer. Am J Clin Pathol. 127:820–827.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Al-Mulla F, Hagan S, Al-Ali W, Jacob SP,
Behbehani AI, Bitar MS, Dallol A and Kolch W: Raf kinase inhibitor
protein: Mechanism of loss of expression and association with
genomic instability. J Clin Pathol. 61:524–529. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li DX, Cai HY, Wang X, Feng YL and Cai SW:
Promoter methylation of Raf kinase inhibitory protein: A
significant prognostic indicator for patients with gastric
adenocarcinoma. Exp Ther Med. 8:844–850. 2014.PubMed/NCBI
|
|
25
|
Minoo P, Baker K, Goswami R, Chong G,
Foulkes WD, Ruszkiewicz AR, Barker M, Buchanan D, Young J and Jass
JR: Extensive DNA methylation in normal colorectal mucosa in
hyperplatic polyposis. Gut. 55:1467–1474. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Connolly R and Stearns R: Epigenetics as a
therapeutic target in breast cancer. J Mammary Gland Biol
Neoplasia. 17:191–204. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu K and Bonavida B: The activated
NF-kappaB-Snail-RKIP circuitry in cancer regulates both the
metastatic cascade and resistance to apoptosis by cytotoxic drugs.
Crit Rev Immunol. 29:241–254. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Umezawa K: Possible role of peritoneal
NF-κB in peripheral inflammation and cancer: Lessons from the
inhibitor DHMEQ. Biomed Pharmacother. 65:252–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Poma P, Notarbartolo M, Labbozzetta M,
Sanguedolce R, Alaimo A, Carina V, Maurici A, Cusimano A, Cervello
M and D'Alessandro N: Antitumor effects of the novel NF-kappaB
inhibitor dehydroxymethyl-epoxyquinomicin on human hepatic cancer
cells: Analysis of synergy with cisplatin and of possible
correlation with inhibition of pro-survival genes and IL-6
production. Int J Oncol. 28:923–930. 2006.PubMed/NCBI
|
|
30
|
Iorio MV and Croce CM: MicroRNAs in
cancer: Small molecules with a huge impact. J Clin Oncol.
27:5848–5856. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fabbri M and Calin GA: Epigenetics and
miRNAs in human cancer. Adv Genet. 70:87–99. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Inguglia L, Notarbartolo M, Poma P,
Labbozzetta M and D'Alessandro N: Induction of apoptosis and
chemosensitization by the histone deacetylase inhibitor
trichostatin A in hepatocellular carcinoma cells: Molecular
analysis and RKIP levels. Forum Immun Dis Ther. 2:127–135. 2011.
View Article : Google Scholar
|
|
33
|
Yang X, Phillips DL, Ferguson AT, Nelson
WG, Herman JG and Davidson NE: Synergistic activation of functional
estrogen receptor (ER)-alpha by DNA methyltransferase and histone
deacetylase inhibition in human ER-alpha-negative breast cancer
cells. Cancer Res. 61:7025–7029. 2001.PubMed/NCBI
|
|
34
|
Zhai FX, Liu XF, Fan RF, Long ZJ, Fang ZG,
Lu Y, Zheng YJ and Lin DJ: RUNX3 is involved in caspase-3-dependent
apoptosis induced by a combination of 5-aza-CdR and TSA in
leukaemia cell lines. J Cancer Res Clin Oncol. 138:439–449. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sethi S, Sarkar FH, Ahmed Q, Bandyopadhyay
S, Nahleh ZA, Semaan A, Sakr W, Munkarah A and Ali-Fehmi R:
Molecular markers of epithelial-to-mesenchymal transition are
associated with tumor aggressiveness in breast carcinoma. Transl
Oncol. 4:222–226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Piao HL, Yuan Y, Wang M, Sun Y, Liang H
and Ma L: α-catenin acts as a tumour suppressor in
E-cadherin-negative basal-like breast cancer by inhibiting NF-κB
signalling. Nat Cell Biol. 16:245–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Walker EJ, Rosenberg SA, Wands JR and Kim
M: Role of raf kinase inhibitor protein in hepatocellular
carcinoma. For Immunopathol Dis Therap. 2:195–204. 2011. View Article : Google Scholar : PubMed/NCBI
|