Introduction
Osteosarcoma is a frequent primary malignant bone
tumor with predilection in children and adolescents (1), the oncogenesis of which has been
reported to be partially associated with transforming growth factor
β (TGF-β) (2). Periosteal
osteosarcoma (POS), an intermediate-grade chondroblastic
osteosarcoma arising from the surface of the long bones, accounts
for <2% of all osteosarcomas (3,4). Previous
studies have demonstrated that certain hereditary diseases
including Rothmund-Thomson syndrome, Bloom syndrome and Li-Fraumeni
syndrome may increase the risk of osteosarcoma (5,6).
Marfan's syndrome (MFS), a rare autosomal dominant
hereditary disorder of connective tissue, primarily affects the
ocular, skeletal and cardiovascular systems, with high penetrance
and variable phenotypes (7). MFS was
reported to be associated with perturbations in TGF-β biology,
which were commonly found to result from mutations in the
fibrillin-1 gene (8,9). One previous study reported the
simultaneous occurrence of MFS and osteosarcoma of the foot in a
patient (10). However, it remains to
be elucidated whether POS is associated with MFS.
The present study reported a case of POS and MSF
co-occurring in a 6-year-old girl, the former of which induced a
pathological femoral shaft fracture. The present report described
the process of the disorders and reviewed the relevant
literature.
Case report
Overview
A 6-year-old girl visited the clinic of Nanfang
Hospital, Southern Medical University (Guangzhou, China) with
aggravating swelling in the right thigh following two previous
surgeries of internal fixation and implant removal, which were
performed due to a femoral shaft fracture in her right leg. The
present study was approved by medical ethics committee of Nanfang
Hospital. Written informed consent from the patient's family was
provided, and the patients' records were anonymized and
de-identified prior to analysis.
Medical history
Abnormal symptoms were denied prior to the
presentation of the fracture in the patient. A blood relationship
between the patient's parents was also denied. The patient's father
had previously received a diagnosis of MFS; however, heredity
disorders were not present in other members of the family.
Initial presentation
Subsequent to a fall during exercise, the patient
presented with pain, swelling, deformity and disability in the
right thigh. Radiograph images performed at a local hospital
revealed a single fracture in the right femoral shaft. The patient
was admitted and received fracture fixation surgery a week later.
Symptomatic treatment and nutritional support were administered
postoperatively. At 2 weeks post surgery, aggravated swelling was
observed and the patient returned to the local hospital. X-ray
examination revealed the formation of an extensive osteotylus in
the affected site. The patient was readmitted and underwent removal
of the implant 1 month following the initial surgery. Aggravated
swelling appeared again, accompanied with superficial venous
engorgement following the second surgery. The patient was then
brought to Nanfang Hospital.
Medical examination
The patient was 123 cm in height and 24 kg in
weight. Physical examination revealed a normal spinal curvature and
a long slender neck. The upper to lower segment ratio was 0.82.
Joint hypermobility was identified in the patient's hands and
fingers. Optic examination showed a slight impairment in both of
the eyes. The circumference at 10 cm above patella of the right
thigh was 52 cm, compared with 31 cm in the left thigh. In
addition, tension blisters as well as superficial venous
engorgement were observed. However, the sensation of the affected
limb was normal.
Laboratory results demonstrated significantly
increased serum levels of white blood cells
(28.73×109/l; normal range, 3.5–9.5×109/l),
C-reactive protein (81.1 mg/l; normal range, 0–5 mg/l) and alkaline
phosphatase (976.4–1100 U/l; normal range, 50–400 U/l). However,
the serum erythrocyte sedimentation rate (21 mm/l) was only
slightly elevated compared with the upper value of normal range (20
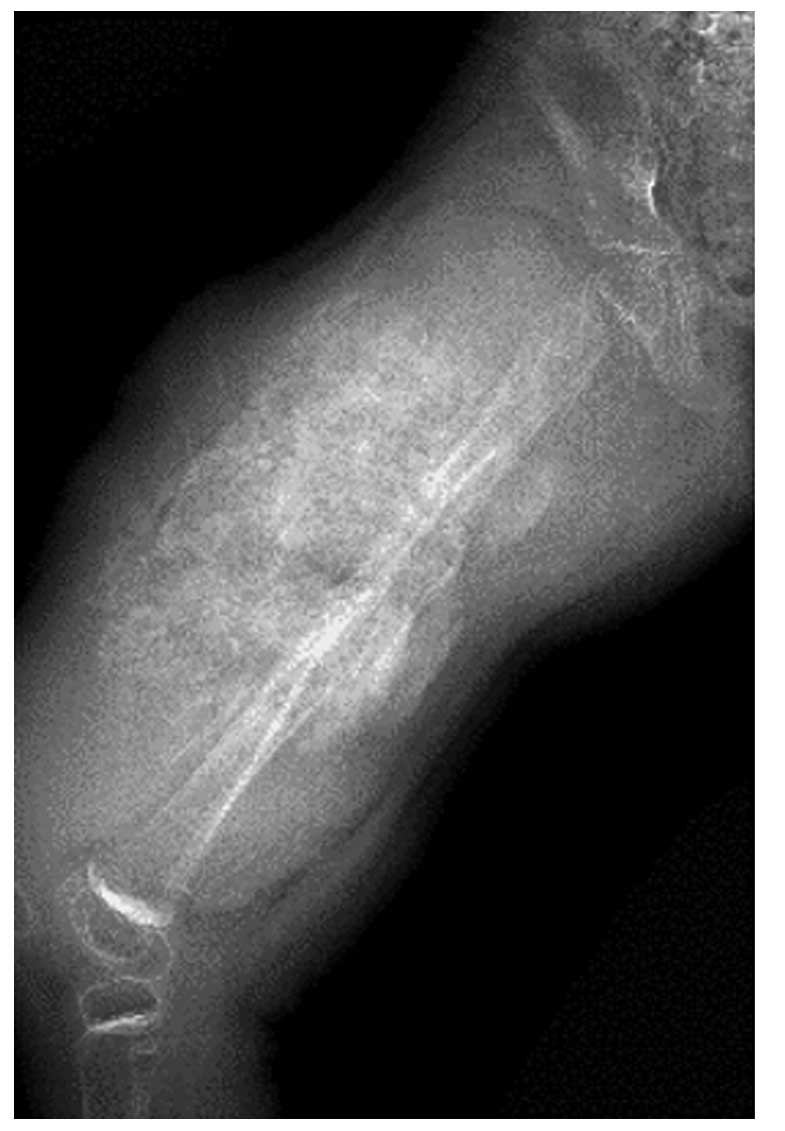
mm/l). Radiograph images revealed extensive osteolytic destruction,
asymmetrical periosteal reaction with Codman's triangle and cortex
thickening, which indicated the possibility of osteosarcoma. No
X-ray abnormities were identified in the hip or the knee (Fig. 1), nor in the chest. Magnetic resonance
imaging (MRI) of right thigh revealed abnormal signals in
T1 and T2-weighted images (Fig. 2A–C). Spinal MRI revealed a biconcave
sign in the partial vertebral bodies (Fig. 2D). An electrocardiogram demonstrated
normal sinus rhythms without other types of arrhythmia. In
addition, an echocardiograph revealed normal sizes and motions of
the ventricles, atria and valves. MFS was diagnosed according to
the aforementioned clinical features and the associated family
history.
Operative procedure and postoperative
follow up
Osteosarcoma was suspected, based on the following
factors: i) Extensive osteotylus was observed <1 month after
fracture; ii) extensive osteolytic destruction, asymmetrical
periosteal reaction with Codman's triangle and cortex thickening
were observed (Fig. 1); and iii)
clinical symptoms and laboratory tests also indicated
abnormalities, including tension blisters and superficial venous
engorgement, as well as signifiantly increased serum levels of
alkaline phosphatase, white blood cells and C-reactive protein.
Thus, radical resection of the tumor was proposed according to the
limb salvage requirement of the patient as well as the patient's
parents. Surgery was performed under general anesthesia. During
surgery, the muscles around the femur were observed to have a
fatty-like change and the aberrant bone grew widely around the bone
shaft. The patient underwent thorough resection of the tumor and
reconstruction. Resected tissues underwent histopathological and
immunohistochemical examinations (Fig. 3A
and B). Histopathological examination revealed marked chondroid
differentiation with hypercellularity and prominent nuclear
pleomorphism. Immunohistochemical examination revealed positive
expression of CD99 (+), osteopontin (+, weakly), Ki-67 (+, 50%),
p53 (+, individually) and S-100 (+), as well as negative expression
of B cell lymphoma 2 (−). Outcomes of the above examinations
confirmed the diagnosis of POS. Postoperative radiographs
demonstrated that the primary tumor was resected completely and the
intramedullary fixation of the femur was in place (Fig. 4).
Discussion
Osteosarcoma is one of the most frequent types of
primary malignant bone tumors, accounting for ~15% of all bone
tumors (11). In addition, the
incidence rate of osteosarcoma arising from the surface of the long
bones is markedly lower compared with those from other sites of the
bone. Surface osteosarcomas are classified into three main types,
including parosteal (juxtacortical), periosteal and high-grade
osteosarcoma (12). POS was first
reported by Ewing (13) in 1939;
subsequently, in 1976, Unni et al (14) described the characteristics of POS
pathology based on 23 patients. As a type of intermediate-grade
osteosarcoma, POS predominantly occurs in males and patients in the
second decade of life (15,16). POS usually arises from the diaphysis
or the meta-diaphyseal surface of the tibia and femur; in addition,
POS is less aggressive and has a better prognosis compared with
other frequent types of osteosarcoma (17,18).
However, controversy still surrounds the treatment of this disease.
The effect of chemotherapy treatment on the prognosis of POS
patients remains indefinite (15),
although it often demonstrates a promising clinical efficacy in the
treatment of osteosarcoma. Grimer et al (15) reported the good prognosis of POS
patients following only resection of the tumor. Thus, the case in
the present study only received resection of the tumor without
chemotherapy or radiotherapy. The role of the two adjunctive
therapy methods in the treatment of POS requires further
investigation.
MFS was first described by Antoine-Bernard Marfan in
1898. MFS is a rare autosomal dominantly inherited systemic
connective tissue disorder with an estimated prevalence of 1–3 per
10,000 (19,20). The disease primarily affects the
cardiovascular, skeletal and ocular systems, with skin, fascia,
lung and adipose tissue occasionally involved (21). Diet et al (22) indicated that MFS was associated with
the genetic defects in the chromosome loci
D15S25 and D15S1. The
disease exhibited a high penetrance and a marked inter- and
intrafamilial variability (23);
however, there remains to be a lack of effective methods for the
treatment of this disorder. Cardiovascular deformities are the
primary fatal factor of MFS; thus, the prevention of
life-threatening cardiovascular complications with MFS is vital.
The main strategies for this include lifestyle modifications,
regular echocardiographic assessment, pharmacological treatment and
prophylactic surgery (24). In the
present study, the patient did not present with any abnormalities
of the cardiovascular system.
To the best of our knowledge, only one previous
study has reported a case of simultaneous osteosarcoma and MFS
(10); in this previous case, the
osteosarcoma was located in the foot. Compared with the previous
case, the present case had several different characteristics.
Initially, the affected site was the femur shaft. Additionally,
fracture was the main reason that accounted for the patient's visit
to the clinic of a local hospital, which treated the patient as a
simple femoral shaft fracture. Subsequently, it was revealed the
patient suffered from the co-occurrence of MFS and POS. The
simultaneous occurrence of these two rare disorders in a patient
may indicate a correlation between MSF and POS. It was previously
reported that heterozygous mutations in TGF-β receptor 2 were
associated with several malignancies and genetic connective-tissue
disorders (25). Previous studies
also indicated that TGF-β had a crucial role in the aortopathy of
MFS (26–28). In addition, one study suggested that
TGF-β was an autocrine factor that regulated the growth of human
osteosarcomas (2). Therefore, it may
be inferred that TGF-β is a potential connection between the
occurrence of MSF and POS.
In conclusion, to the best of our knowledge, the
present study was the first to report a case of simultaneous POS
and MFS in a child. The case report identified the characteristics
of POS and key points of the disease in the process of diagnosis
and treatment. The present study may therefore increase the
awareness of orthopedists on the possibility of malignant bone
tumor in a hereditary patient with osteotylus overgrowth following
fracture surgery. In addition, due to the simultaneous appearance
of POS and MFS in the present case, it was suggested that further
studies should be conducted to determine whether there is an
association between the two rare disorders.
Acknowledgements
The authors thank Professor Allen P. Liang for
revising and editing this manuscript.
References
|
1
|
Durnali A, Alkis N, Cangur S, Yukruk FA,
Inal A, Tokluoglu S, Seker MM, Bal O, Akman T, Inanc M, et al:
Prognostic factors for teenage and adult patients with high-grade
osteosarcoma: An analysis of 240 patients. Med Oncol. 30:6242013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Franchi A, Arganini L, Baroni G, Calzolari
A, Capanna R, Campanacci D, Caldora P, Masi L, Brandi ML and Zampi
G: Expression of transforming growth factor beta isoforms in
osteosarcoma variants: Association of TGF beta 1 with high-grade
osteosarcomas. J Pathol. 185:284–289. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cesari M, Alberghini M, Vanel D, Palmerini
E, Staals EL, Longhi A, Abate M, Ferrari C, Balladelli A and
Ferrari S: Periosteal osteosarcoma: A single-institution
experience. Cancer. 117:1731–1735. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maheshwari AV, Jelinek JS, Seibel NL,
Meloni-Ehrig AM, Kumar D and Henshaw RM: Bilateral synchronous
tibial periosteal osteosarcoma with familial incidence. Skeletal
Radiol. 41:1005–1009. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fuchs B and Pritchard DJ: Etiology of
osteosarcoma. Clin Orthop Relat Res. 40–52. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Malkin D, Li FP, Strong LC, Fraumeni JF
Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ and Tainsky
MA: Germ line p53 mutations in a familial syndrome of breast
cancer, sarcomas and other neoplasms. Science. 250:1233–1238. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Summers KM, Xu D, West JA, et al: An
integrated approach to management of Marfan syndrome caused by an
FBN1 exon 18 mutation in an Australian Aboriginal family. Clin
Genet. 65:66–69. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pearson GD, Devereux R, Loeys B, et al:
Report of the national heart, lung and blood institute and national
marfan foundation working group on research in Marfan syndrome and
related disorders. Circulation. 118:785–791. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dietz HC, Cutting GR, Pyeritz RE, Maslen
CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ
and Curristin SM: Marfan syndrome caused by a recurrent de novo
missense mutation in the fibrillin gene. Nature. 352:337–339. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roopnariane A, Freed RJ, Price S, Fox EJ
and Ritty TM: Osteosarcoma in a Marfan patient with a novel
premature termination codon in the FBN1 gene. Connect Tissue Res.
52:157–165. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murphey MD, Robbin MR, McRae GA, Flemming
DJ, Temple HT and Kransdorf MJ: The many faces of osteosarcoma.
Radiographics. 17:1205–1231. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schajowicz F, McGuire MH, Santini Araujo
E, Muscolo DL and Gitelis S: Osteosarcomas arising on the surfaces
of long bones. J Bone Joint Surg Am. 70:555–564. 1988.PubMed/NCBI
|
|
13
|
Ewing J: A review of the classification of
bone tumours. Bull Am Coll Surg. 24:290–295. 1939.
|
|
14
|
Unni KK, Dahlin DC and Beabout JW:
Periosteal osteogenic sarcoma. Cancer. 37:2476–2485. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grimer RJ, Bielack S, Flege S, Cannon SR,
Foleras G, Andreeff I, Sokolov T, Taminiau A, Dominkus M,
San-Julian M, et al: Periosteal osteosarcoma-a European review of
outcome. Eur J Cancer. 41:2806–2811. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ritts GD, Pritchard DJ, Unni KK, Beabout
JW and Eckardt JJ: Periosteal osteosarcoma. Clin Orthop Relat Res.
219:299–307. 1987.PubMed/NCBI
|
|
17
|
Hall RB, Robinson LH, Malawar MM and
Dunham WK: Periosteal osteosarcoma. Cancer. 55:165–171. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lim C, Lee H, Schatz J, Alvaro F, Boyle R
and Bonar SF: Case report: Periosteal osteosarcoma of the clavicle.
Skeletal Radiol. 41:1011–1015. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gray JR, Bridges AB, Faed MJ, Pringle T,
Baines P, Dean J and Boxer M: Ascertainment and severity of Marfan
syndrome in a Scottish population. J Med Genet. 31:51–54. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Judge DP and Dietz HC: Marfan's syndrome.
Lancet. 366:1965–1976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Loeys BL, Dietz HC, Braverman AC,
Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y,
Jondeau G, Faivre L, Milewicz DM, et al: The revised Ghent nosology
for the Marfan syndrome. J Med Genet. 47:476–485. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dietz HC, Pyeritz RE, Hall BD, Cadle RG,
Hamosh A, Schwartz J, Meyers DA and Francomano CA: The Marfan
syndrome locus: Confirmation of assignment to chromosome 15 and
identification of tightly linked markers at 15q15-q21.3. Genomics.
9:355–361. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pyeritz RE: The Marfan syndrome. Annu Rev
Med. 51:481–510. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cook JR and Ramirez F: Clinical,
diagnostic and therapeutic aspects of the Marfan syndrome. Adv Exp
Med Biol. 802:77–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizuguchi T, Collod-Beroud G, Akiyama T,
Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M,
Morisaki H, et al: Heterozygous TGFBR2 mutations in Marfan
syndrome. Nat Genet. 36:855–860. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mizuguchi T and Matsumoto N: Recent
progress in genetics of Marfan syndrome and Marfan-associated
disorders. J Hum Genet. 52:1–12. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sawaki D and Suzuki T: Targeting
transforming growth factor-β signaling in aortopathies in Marfan
syndrome. Circ J. 77:898–899. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Agg B, Benke K, Szilveszter B, Pólos M,
Daróczi L, Odler B, Nagy ZB, Tarr F, Merkely B and Szabolcs Z:
Possible extracardiac predictors of aortic dissection in Marfan
syndrome. BMC Cardiovasc Disord. 14:472014. View Article : Google Scholar : PubMed/NCBI
|