Introduction
Lung cancer is the leading cause of
cancer-associated mortality in China (1). Despite advances in diagnostic and
therapeutic modalities against lung cancer, there has been little
improvement in patient prognostic outcomes. A clinical study
identified immunotherapy as a potentially favorable alternative
approach to the treatment of lung cancer (2). A large number of tumor-associated
antigens have been identified in various types of human cancer,
including the cancer-testis (CT) antigens. CT antigens are of
significant interest as they are expressed in a variety of
malignant neoplasms, normal testis germ cells and the placenta, but
not in other normal human tissues (3). Melanoma-associated antigen A4 (MAGE-A4)
is a CT antigen that has been reported to be expressed in
melanomas, germ cell tumors, certain sarcomas and lung cancer
(4). Patients with MAGE-A4-expressing
tumors are capable of exhibiting specific cellular and humoral
immune responses to targeting MAGE-A4 (5). However, while an abundant infiltration
of CD8+ T cells has frequently been reported to improve
clinical outcomes (6), increased
tumor MAGE-A4 expression is associated with poor survival in
patients with lung cancer (7,8). This likely results from the inhibition
of tumor-specific immune responses in patients with cancer, and
while a number of complex factors are implicated in this process
(9), myeloid-derived suppressor cells
(MDSCs) have a prominent role (10,11). MDSCs
are a heterogeneous group of pathologically activated immature
myeloid cells and myeloid precursors that possess potent
immunosuppressive activity (10–12). MDSCs
have been identified in the peripheral blood, lymphoid tissue and
tumor tissue in a number of experimental mouse models (13). Other studies have also demonstrated
that MDSCs inhibit the effector function of T cells in
tumor-bearing animals and patients with cancer (14,15). The
association between infiltration of CD8+ T cells and
increased tumor MAGE-A4 expression is not clear. Abundant
infiltration of CD8+ T cells has frequently been
reported to improve clinical outcomes. Clinical evidence also
confirmed that MAGE-A4-expressing tumors are capable of exhibiting
specific cellular and humoral immune responses to targeting
MAGE-A4. However, increased tumor MAGE-A4 expression is associated
with poor survival in patients with lung cancer (16).
Therefore, the present study aimed to identify the
reason behind this association. The expression of MAGE-A4 was
evaluated and the number of circulating MDSC were determined in a
mouse model of Lewis lung cancer (LLC), to aid the elucidation of
the association between MDSCs and MAGE-A4.
Materials and methods
Cell lines and animals
The LLC tumor cell line was purchased from the
American Type Culture Collection (ATCC® CRL-1642™; Manassas, VA,
USA) and maintained in the Department of Immunology, Peking Union
Medical College and Chinese Academy of Medical Sciences (Beijing,
China). The B16F10 melanoma control cells were provided by Dr
Lieping Chin (Department of Immunology, School of Medicine, Yale
University, New Haven, CT, USA) (17). These murine tumor cells were
maintained in Dulbecco's modified Eagle's medium/F12 medium
containing 10% heated-inactivated fetal calf serum (FCS), 2 mM
L-glutamine, 10 mM HEPES, penicillin (100 U/ml)-streptomycin (50
µg/ml) solution and 1% sodium pyruvate solution (Gibco Life
Technologies, Carlsbad, CA, USA).
Female 6–8 week-old C57BL/6 mice (n=20) were
obtained from the Experimental Animal Institute of Peking Union
Medical College. Mice were subjected to a 12 h light/dark cycle and
maintained in a specific pathogen-free environment at 18–23°C with
40–60% humidity. Food and water were accessible at all times. All
protocols involving animals used in the present study were approved
by the Animal Research Ethics Committee of the Cancer Institute and
Hospital, Peking Union Medical College and Chinese Academy of
Medical Sciences (no. 20140002, Beijing, China).
In vivo depletion of MDSC
LLC tumor cells (1×106) were resuspended
in 100 µl phosphate-buffered saline (PBS) and subcutaneously
inoculated into the right flanks of four C57BL/6 mice. These LLC
tumor-bearing mice were enrolled in the study when the primary
tumor reached 4–8 mm in diameter. The monoclonal antibody (mAb)
Gr-1 (RB6–8C5; 200 µg in 100 µl PBS; BioLegend, Inc., San Diego,
CA, USA) was intraperitoneally administered on days 5, 10 and 15 to
deplete MDSCs. Control LLC tumor-bearing mice (n=1) were treated
with immunoglobulin G (rat IgG2b; BioLegend, Inc.).
Tumor dimensions were measured two or three times per week with
digital calipers (Cangzhou Yongkang Medical Devices Co., Ltd.,
Yongkang, China), and the tumor volume was calculated using the
formula: Tumor volume (mm3) = 0.52 × (length ×
width2). Overall animal survival was monitored following
conclusion of the treatment. Subsequently, mice were sacrificed 19
days after tumor cell inoculation, using carbon dioxide and tumor
tissues were resected. A total of 5 mice were used per group, and
all experiments were repeated twice. All experiments were conducted
according to animal studies ethics committee guidelines.
Flow cytometric analysis
Cells from the bone marrow or spleen of LLC
tumor-bearing mice were harvested 19 days after tumor cell
inoculation, under sterile conditions. Single-cell suspensions were
prepared, and red blood cells were removed using Tris lysis buffer
(144 mM NH4Cl, 17 mM Tris-HCl; pH 7.2; Zhongshan Golden
Bridge Biotechnology Co., Ltd., Beijing, China). Mononuclear cells
isolated from the bone marrow or spleen were incubated at 4°C for
30 min with the following fluorescently conjugated rat mAbs
purchased from BioLegend, Inc. [0.2 µg Abs per 106 cells
(100 µl)]: Anti-CD45 (allophycocyanin; 30-F11), anti-CD11b
[fluorescein isothiocyanate (FITC); M1/70], anti-Gr-1
[phycoerythrin (PE), RB6–8C5], anti-CD8 (PE; 53–6.7) and anti-CD3
(PE/Cy7; 145-2C11). Subsequently,
CD45+/CD11b+/Gr-1+ cells (MDSCs)
or CD45+/CD3+/CD8+ cells were
purified by cell sorting using a FACS Calibur (BD Biosciences,
Franklin Lakes, NJ, USA) instrument to >90% purity.
MDSC culture was performed as described previously
with modifications (18). Briefly,
sorted bone marrow populations (CD11b+/Gr-1+
and CD3+/CD8+) were cultured in six-well
plates (1×105 cells/well) for 3 days with the addition
of recombinant mouse granulocyte-macrophage colony-stimulating
factor (GM-CSF; 50 ng/ml; R&D Systems, Minneapolis, MN, USA),
prior to harvesting the supernatant for use. CD8+ T
cells obtained from the spleen were used as effector cells for a
cytotoxic assay.
For surface staining, murine leukocytes from
heparinized whole tail blood (40 µl) collected on days 8 and 19
post-tumor challenge (n=4) were incubated with the following
antibodies: Anti-CD45, anti-CD11b, anti-Gr-1, anti-CD8, anti-CD4
and anti-CD3 (BioLegend, Inc.), at a concentration of 0.2 µg Abs
per 106 cells (100 µl) at 4°C for 30 min, according to
standard protocols (19).
Subsequently, 500 µl OptiLyse® lysis solution (Beckman Coulter,
Brea, CA, USA) was added to each tube and incubated at room
temperature for 10 min. Following washing with PBS, the samples
were subjected to flow cytometric analysis. Additional analysis was
performed using FlowJo software (version 7.6.5; Tree Star, Inc.,
Ashland, OR, USA).
Cell proliferation assay
LLC cells were incubated with 5 µM
5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE;
Invitrogen Life Technologies, Carlsbad, CA, USA) at 37°C for 10 min
in serum-free media, and then washed in RPMI-1640 media containing
10% FCS (Gibco Life Technologies). CFSE-labeled cells were cultured
in complete media with the addition of supernatant from MDSCs in
24-well plates (1 ml/well, complete media and supernatant ratio,
1:1). Following 30 h of incubation at 37°C and 5% CO2,
the number of cell divisions undertaken by LLC cells was determined
by flow cytometry.
Immunoblot analysis
Protein was extracted from LLC tumor tissues from
C57BL/6 tumor-bearing mice, as well as LLC and B16F10 tumor cells,
and analyzed by western blotting. Briefly, tumor tissues from
C57BL/6 mice were flash-frozen in liquid nitrogen immediately
following collection, while LLC or B16F10 tumor cells were
harvested and washed with PBS. Ground tumor tissues and LLC or
B16F10 cells were then lysed in pre-chilled
radioimmunoprecipitation assay lysis buffer (Thermo Fisher
Scientific, Waltham, MA, USA) containing protease and phosphatase
inhibitor cocktails (Roche Diagnostics, New Providence, NJ, USA).
Following centrifugation at 10,000 × g for 15 min at 4°C, the
supernatant was removed and protein concentration determined using
Coomassie brilliant blue G-250 (Sigma-Aldrich, St. Louis, MO, USA).
Total protein (20 µg) was prepared for denaturing gel
electrophoresis. For western blotting, proteins were separated on
12% SDS-PAGE gels (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and transferred onto Immobilon-FL polyvinylidene difluoride
membranes (EMD Millipore, Billerica, MA, USA) using a wet transfer
apparatus (Bio-Rad Laboratories, Inc.). Following transfer, the
blots were rinsed in washing buffer [Tris-buffered saline with
Tween-20 (TBST): 20 mM Tris base, 137 mM NaCl, 0.1% Tween-20, pH
7.6] and blocked in blocking buffer (5% non-fat dry milk in TBST;
both from Bio-Rad Laboratories, Inc.) for 1 h at room temperature.
Membranes were then washed in TBST and incubated with primary
antibodies (1:1,000; Cell Signaling Technology, Inc., Danvers, MA,
USA) diluted in TBST with 5% bovine serum albumin overnight at 4°C.
Subsequent washes in TBST were followed by incubation with
secondary antibodies [goat anti-rabbit IgG-horseradish peroxidase
(HRP), cat. no. 7074, or goat anti-mouse IgG-HRP, cat. no. 7076;
1:2,000; Cell Signaling Technology, Inc.] diluted in blocking
buffer for 1 h at room temperature. Subsequently, the membranes
were washed in TBST and the protein bands were visualized using an
enhanced chemiluminescence western blotting system (GE Healthcare
Life Sciences, Piscataway, NJ, USA). GAPDH and β-actin were used as
internal controls for consistent protein loading. The following
primary antibodies were used: Rabbit polyclonal MAGE-A4 antibody
(AV54410), rabbit phosphorylated (p)-AKT473 XP® mAb
(Ser473; D9E; 4060), rabbit total (t)-AKT mAb (pan; C67E7; 4691),
rabbit p-signal transducer and activator of transcription 3
(STAT3)705 XP® mAb (Tyr705; D3A7; 9145), rabbit
p-STAT3727 mAb (Ser727; 9134), rabbit t-STAT3 mAb (79D7;
4904), mouse p-extracellular signal-regulated kinase (ERK)1/2 mAb
[p44/42 MAPK (Erk1/2); Thr202/Tyr204; E10; 9106], rabbit t-ERK1/2
mAb [p44/42 MAPK (Erk1/2); 9102], mouse β-actin mAb (8H10D10;
12262) and rabbit GAPDH mAb (14C10; 2118). MAGE-A4 rabbit
polyclonal antibody was purchased from Sigma-Aldrich, while all
other primary antibodies mentioned above were obtained from Cell
Signaling Technology, Inc. NuPAGE® LDS Sample Buffer (NP0007),
NuPAGE® MOPS SDS Running Buffer (NP0001-02) and Novex® Tris-Glycine
Transfer Buffer (LC3675) for western blotting were purchased from
Invitrogen Life Technologies. The intensities of bands on the
membranes were determined using Quantity One software version 4.6.2
(Bio-Rad, Laboratories, Inc., Hercules, CA, USA).
Cytotoxicity assay
LLC tumor cells (target cells) were labeled with 5
µM CFSE. Sorted CD8+ T cells were used as effector
cells. Effector cells (4×105 cells/well) were cultured
in 48-well plates with 2×104 labeled tumor target
cells/well in 500 µl RPMI-1640 containing 10% FCS. Following 4 h of
incubation, propidium iodide (PI; 2 µg/ml, Sigma-Aldrich) was added
to identify killed target cells. Flow cytometric analysis
identified PI+CFSE+ cells (killed targets)
and PI−CFSE+ cells (viable targets). A total
of ~5,000 target cells were acquired. The percentage of specific
lysis was calculated in relation to the proportion of basal lysis
in untreated cells using the following formula: Percentage of
specific lysis = proportion of lysis per sample - proportion of
basal lysis / (1 - proportion of basal lysis) × 100.
Statistical analysis
GraphPad Prism V (GraphPad Software Inc., La Jolla,
CA, USA) was used for statistical analyses. Data are presented as
the mean ± standard error of the mean, unless indicated otherwise.
For comparisons between two groups, statistical analyses were
performed using a Student's t-test, and P<0.05 was considered to
indicate a statistically significant difference. Survival curves
were estimated using the Kaplan-Meier method, and differences among
these were evaluated using the log-rank test.
Results
MDSCs regulate the expression of
MAGE-A4 via the p-STAT3705 pathway
To establish a mouse model of lung cancer, which
highly expressed the CT antigen MAGE-A4, MAGE-A4 expression was
first detected in LLC tumor cells. LLC tumor cells highly expressed
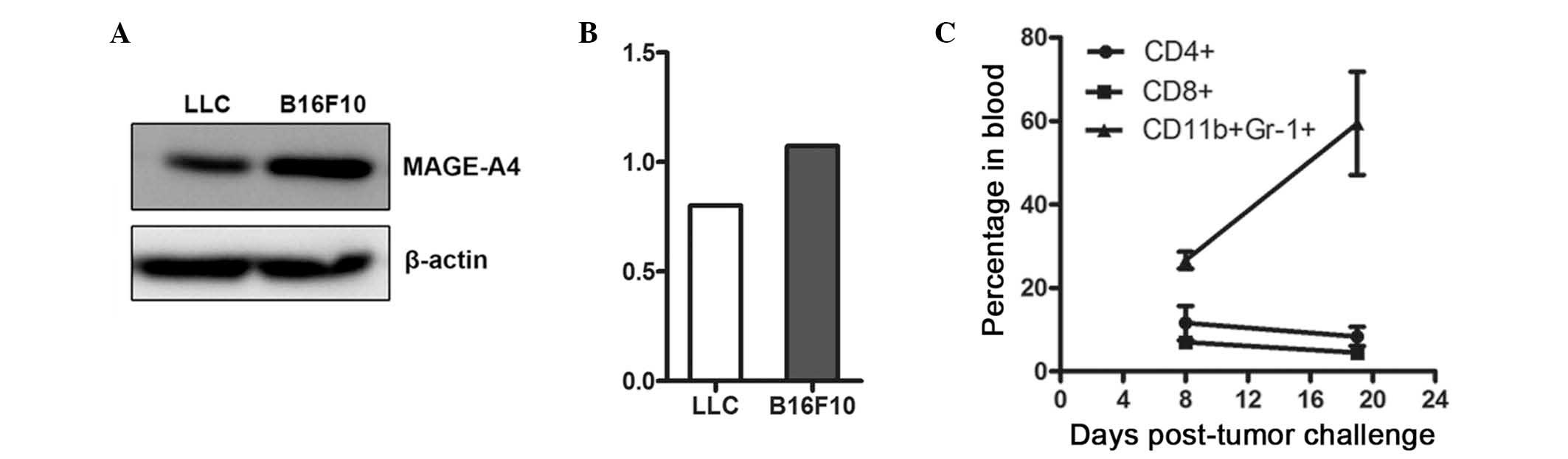
the MAGE-A4 antigen, as did positive control B16F10 cells (Fig. 1A and B). Subsequently, the number of
MDSCs present in the blood of tumor-bearing animals at various
stages of tumor development was determined by flow cytometry, and
an increased MDSC population was identified in advanced tumors
(Fig. 1C).
To investigate the impact of MDSCs on MAGE-A4
expression, MDSCs were depleted in LLC tumor-bearing animals.
Western blotting revealed that targeted depletion of MDSCs
decreased MAGE-A4 expression in tumor-bearing mice. To elucidate
the mechanism underlying how MDSCs enhanced MAGE-A4 expression in
LLC tumor-bearing mice, p-AKT473, t-AKT,
p-STAT3705, p-STAT3727, t-STAT3, p-ERK1/2 and
t-ERK1/2 expression were evaluated by western blotting. The results
presented in Fig. 2A revealed that
depletion of MDSCs significantly inhibited MAGE-A4 expression, and
that this effect was associated with the p-AKT473 and
p-STAT3705 pathways.
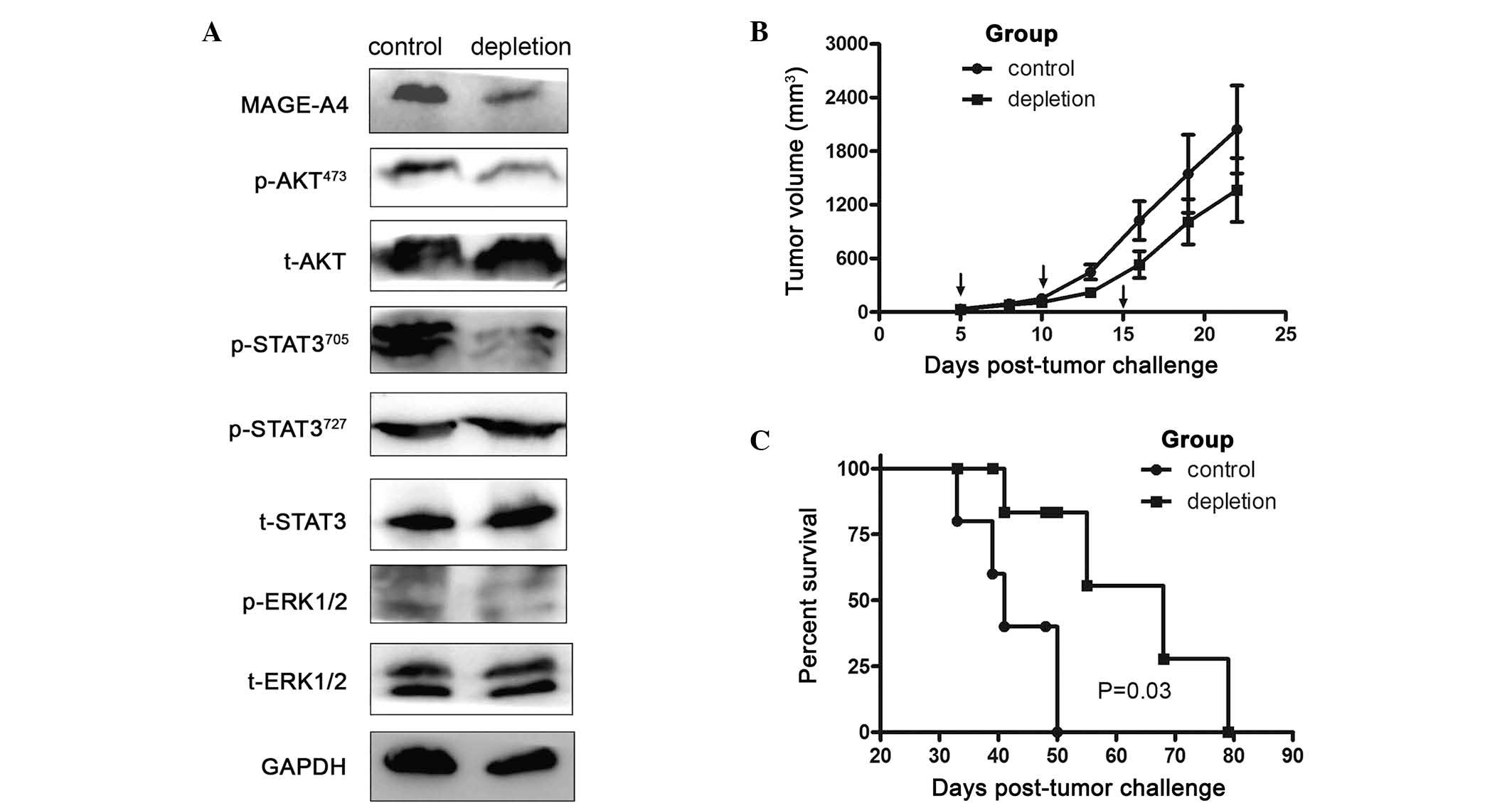 | Figure 2.Depletion of MDSCs decreases MAGE-A4
expression and prolongs survival of tumor-bearing mice. (A) Protein
was isolated from tumor tissues 19 days subsequent to tumor cell
inoculation. MAGE-A4, p-AKT473, t-AKT,
p-STAT3705, p-STAT3727, t-STAT3, p-ERK1/2 and
t-ERK1/2 levels in the tumor tissue were evaluated by western
blotting. GAPDH levels served as an internal control. (B) LLC tumor
cells (1×106) in 100 µl phosphate-buffered saline were
subcutaneously inoculated into the right flanks of C57BL/6 mice.
Tumor growth curves compare subcutaneous tumor growth in the
control and MDSC depletion groups, determined by caliper
measurement. Arrows indicate the times of depletion assay. Values
are expressed as the mean ± standard error of the mean. (C)
Kaplan-Meier estimate of overall survival, comparing control and
MDSC depletion groups. There was a statistically significant
difference between the two groups, P=0.03. LLC, Lewis lung cancer;
MAGE-A4, melanoma-associated antigen A4; MDSCs, myeloid-derived
suppressor cells; p, phosphorylated; STAT3, signal transducer and
activator of transcription 3; ERK, extracellular signal-regulated
kinase; t, total. |
Depletion of MDSCs prolongs survival
of mice bearing LLC tumors
The survival of mice bearing LLC tumors following
depletion of MDSCs was subsequently evaluated. Tumor masses did not
generally decrease in size during the treatment period (Fig. 2B). However, depletion of
CD11b+Gr1+ cells conferred a significant
survival advantage, with median survival of 68 days, whereas all
control mice treated with control isotype IgG succumbed by day 50
(median survival, 41 days; Fig. 2C).
Although this result may be interpreted as indicating a lack of
efficacy, acute increases in tumor size following immune therapy
have frequently been attributed to the recruitment and infiltration
of immune cells, rather than the progressive growth of cancer cells
(20).
Depletion of MDSCs enhances
CD8+ T cell accumulation
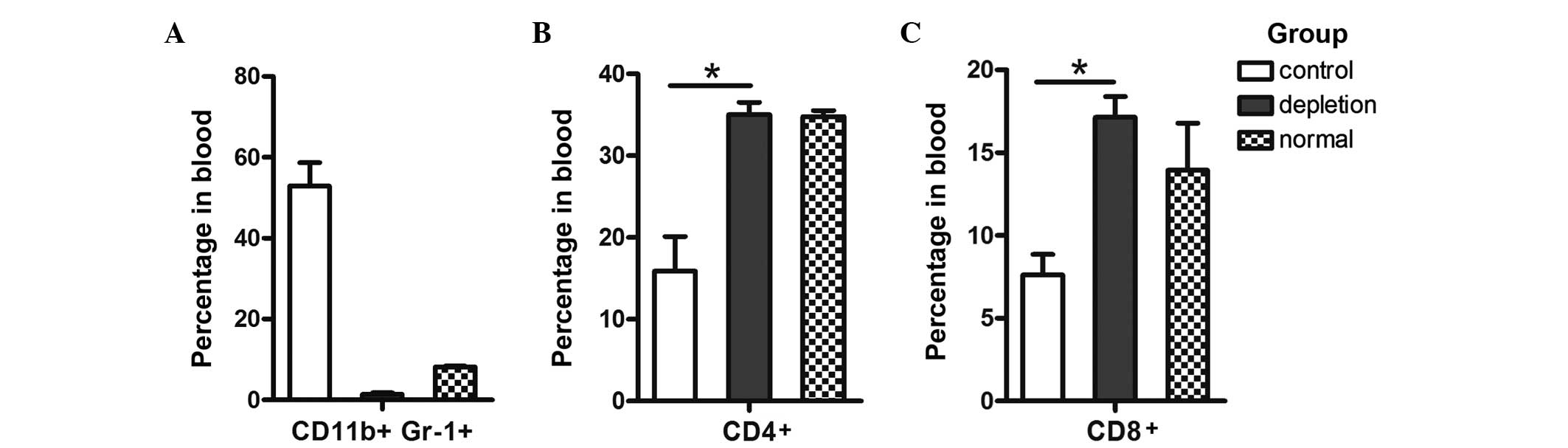
Administration of the mAb Gr-1 markedly reduced the
percentage and number of circulating MDSCs in LLC tumor-bearing
C57BL/6 mice (Fig. 3A). The
percentage of circulating CD4+ T cells increased ~2-fold
(13.11±2.59 in control mice vs. 23.5±1.3 in RB6–8C5-treated mice;
n=4 per group; P=0.053; Fig. 3B);
however, this result was not statistically significant. Consistent
with this trend, the percentage of circulating CD8+ T
cells (Fig. 3C) in RB6–8C5-treated
mice increased ~2-fold when compared with that of untreated mice,
and this result was statistically significant (7.63±1.23 in control
mice vs. 17.15±1.25 in RB6–8C5-treated mice; n=4 per group;
P=0.032).
MDSCs suppress the cytotoxic activity
of CD8+ T cells
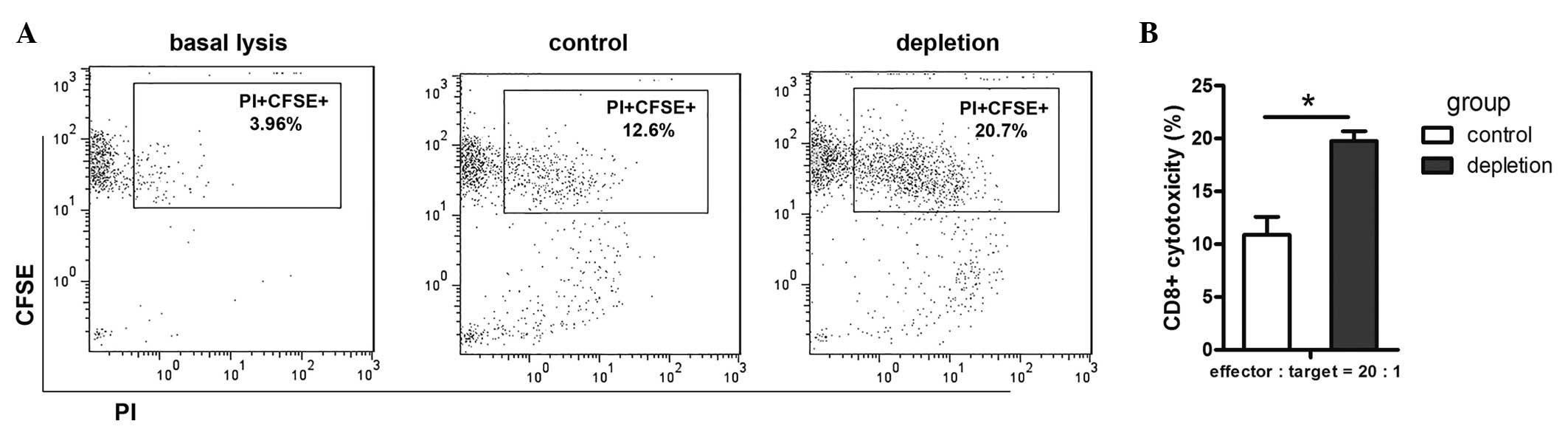
To evaluate the effect of MDSCs on the efficacy of
the cytotoxic immune response in the LLC animal model,
CD8+ T cell cytotoxic activity was assessed by flow
cytometry. CD8+ T cell cytotoxicity towards LLC cells
was found to be higher in the MDSC depletion group than that of the
control group (Fig. 4A and B). This
confirmed that MDSC depletion in tumor-bearing mice resulted in
functional activation of CD8+ T cells, which, in
combination with other factors, results in tumor regression.
These results demonstrated that cancer-conditioned
MDSCs may impede the recognition of tumor antigens by
CD8+ T cells and impair effector T cell activity in
tumor-bearing mice. Furthermore, targeted depletion of MDSCs with
Gr-1 therapy may enhance the endogenous anti-tumor immune
response.
MDSC culture supernatant promotes
proliferation of LLC tumor cells and enhances MAGE-A4
expression
The correlation between MAGE-A4 antigen expression
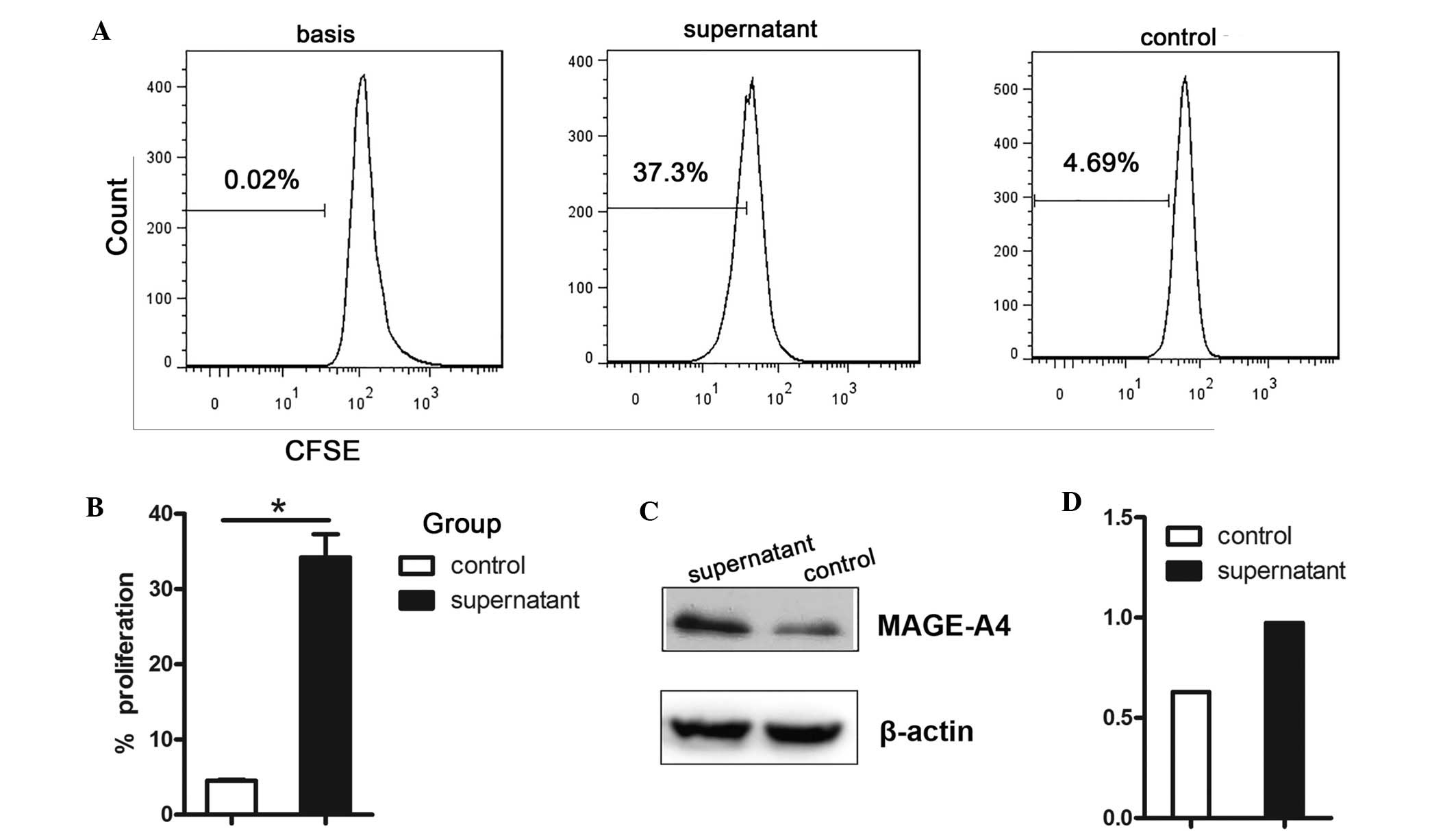
and MDSC infiltration was further assessed by incubating LLC tumor
cells with MDSC supernatant. MDSC supernatant significantly
enhanced LLC cell proliferation (Fig. 5A
and B; P=0.011) and MAGE-A4 expression (Fig. 5C and D).
Discussion
The present study aimed to address the integrated
association between MAGE-A4 expression, MDSC accumulation and
clinicopathological characteristics including overall survival, as
well as circulating CD4+ and CD8+ T cell
levels in LLC tumor-bearing C57BL/6 mice. MAGE-A4 was selected for
investigation due to its potential to elicit marked immune
reactions (21). MAGE-A4 is a member
of the CT antigen family and is not expressed in normal tissues,
other than the testis and placenta. Additionally, high levels of
MAGE-A4 expression are detected in lung cancer (22). This suggests that MAGE-A4 may be an
optimal therapeutic target candidate, and the generation of an
immune response to MAGE-A4 has previously been investigated
(23). In addition, tumor-specific
cytotoxic T lymphocytes (CTLs) recognizing MAGE gene products have
been reported, and a large number of CTL epitopes within MAGE
proteins have been identified (3,24–26). Furthermore, CTL epitopes within
MAGE-A4 have been found to be presented by MHC class I (27–29), and
an intensive accumulation of CD8+ T cells in the tumor
nest has been reported to be correlated with favorable patient
prognosis in numerous types of tumor (30–32).
Theoretically, high expression levels of MAGE-A4 antigen should
induce CTLs to infiltrate the tumor site and result in a favorable
prognosis. However, in the present study, MAGE-A4 expression was
associated with poor survival in tumor-bearing mice. A similar
inverse correlation between MAGE-A4 expression and patient survival
has been previously reported (2,3,33,34). The
precise reasons underlying why higher expression of MAGE-A4 does
not improve patient prognosis have remained elusive. However,
investigation of the accumulation of MDSCs in tumor-bearing
individuals may aid the elucidation of the mechanisms underlying
this effect. To the best of our knowledge, the present study was
the first to report that depletion of a single immunosuppressive
myeloid cell subset (CD11b+Gr-1+) reduces
MAGE-A4 antigen expression via the p-AKT and p-STAT3705
pathways, and induces endogenous CD4+ and
CD8+ T cell accumulation in LLC tumor-bearing mice.
The term ‘cancer’ implies at least a certain degree
of failure of immunity. Since tumors are self-derived, they benefit
from multiple mechanisms of self-tolerance, including immune
evasion (35). Neoplastic cells have
additional elaborate mechanisms to subvert antitumor T cell
activity, for example the induction of host immunosuppressive
cells, including MDSCs and regulatory T cells, and MDSCs have been
identified in a variety of human malignancies (36). Immunosuppression is not limited to the
tumor microenvironment, and circulating myeloid cells capable of
inducing dysfunctional immune responses have been repeatedly
described (37,38). Therefore, the levels of circulating
MDSCs were evaluated, to determine their effect on MAGE-A4
expression. It was demonstrated that MAGE-A4 expression was
upregulated with tumor development, and downregulated with
depletion of MDSCs in LLC tumor-bearing mice. However, no direct
association was identified between MDSCs and carcinogenesis or
other molecules that affect the expression of MAGE-A4 in LLC
tumor-bearing mice.
The number of MDSCs in the circulation exhibited an
exclusive association with MAGE-A4 expression, and may also be
correlated with prognosis. Evidence supporting a role for MDSCs in
modulating MAGE-A4 expression is provided by the results of the
assessment of LLC tumor cell proliferation, which demonstrated that
MDSC supernatant was able to promote the growth of tumor cells and
induce increased expression of MAGE-A4. MDSCs secrete multiple
factors that may support the growth and survival of tumor cells
(39–42), and it would be of interest to confirm
whether MDSCs secrete soluble factors that regulate MAGE-A4
expression. In further support of this, it has previously been
reported that CT antigen expression is association with cell cycle
progression and proliferation (43–45),
apoptosis (13) and susceptibility of
cancer cells to cytokines (46),
suggesting that the CT antigen itself may be associated with
prognosis.
It is possible that a therapeutic strategy whereby
CTLs targeting MAGE-A4 antigens may improve patient outcome
(30). However, the present results
demonstrated that the survival of tumor-bearing mice was prolonged
following depletion of MDSCs, and the subsequent decrease in
MAGE-A4 expression. Therefore, delineation of the mechanisms
underlying MDSC expansion and enhanced MAGE-A4 expression in cancer
represents an area of significant potential. A number of mechanisms
may be exploited to disrupt the immunosuppressive capabilities of
MDSCs, including impairing their trafficking to tumors, preventing
accumulation, limiting immune cell activation (47) or inhibiting their recognition of
tumor-associated antigens, including MAGE-A4. The present results
demonstrated that the endogenous immune response does not have the
capacity to recognize MAGE-A4 antigens, and that the MDSCs
represent a critical barrier to eliciting such immune activity.
The present study indicated that depletion of MDSCs
resulted in decreased expression of MAGE-A4, and that the induction
of specific CTLs targeting MAGE-A4 decreased. Given that MAGE-A4
expression is regulated by MDSCs via the p-STAT3705
pathway, it is possible that targeted depletion of MDSCs, while
simultaneously enhancing the expression of MAGE-A4 via activation
of the p-STAT3705 pathway, may provide the opportunity
to induce an activated CD8+ T cell response. Therefore,
combining CT antigen therapy with targeted disruption of MDSCs and
additional immune-based strategies may help to harness the full
potential of the immune system to recognize and eradicate
malignancies.
It is also possible that other cells and cytokines
in the LLC environment, for example regulatory T cells and
interleukin-6 or transforming growth factor-β (which may be induced
by MDSCs), may also contribute to the enhanced expression of
MAGE-A4 identified in LLC (22).
Further studies are required to elucidate the direct and indirect
regulation of MAGE-A4 expression by MDSCs.
An ideal immunotherapeutic strategy for lung cancer
would maximize the therapeutic index by improving antitumor
effector immune cell functions, while inhibiting immune suppressor
cells. The poor prognosis associated with MAGE-A4-expressing tumors
highlights the need for the development of an aggressive therapy
for their treatment. On the other hand, thesignificant correlation
between MAGE-A4 expression and MDSC accumulation observed in the
present study suggests that the spontaneous immune response may be
incapable of overcoming their immunosuppressive state to eradicate
the established tumor. These findings therefore indicate a novel
mechanism of immunosuppression in the tumor microenvironment and
provide a clear rationale for targeting MDSCs to enhance
immune-based treatment or endogenous immune recognition of
cancer.
In conclusion, to the best of our knowledge, the
present study provides the first report of an association between
MAGE-A4 and MDSCs. The data revealed that MDSCs present within the
tumor may function as immunoregulators, which modulate cell
signaling pathways in the tumor microenvironment. Additionally, the
results demonstrated a critical role for the p-STAT3705
pathway as a key mediator of immune suppressor cell differentiation
(48) and LLC survival. Therefore,
immunotherapeutic strategies involving induction of CTLs to target
MAGE-A4, in combination with MDSC depletion, may provide an
effective approach to cancer immunotherapy.
Acknowledgements
The present study is a project of the Shandong
Province Higher Educational Science and Technology Program (no.
J13LK58) and was supported in part by a Grant-in-Aid from the Zibo
Vocational Institute (no. 2013VC01).
References
|
1
|
Zhi X: The Sixth South-North forum of lung
cancer in China was held in CPPCC auditorium. Zhongguo Fei Ai Za
Zhi. 16:661–662. 2013.(In Chinese). PubMed/NCBI
|
|
2
|
Shigematsu Y, Hanagiri T, Shiota H, Kuroda
K, Baba T, Mizukami M, So T, Ichiki Y, Yasuda M, So T, et al:
Clinical significance of cancer/testis antigens expression in
patients with non-small cell lung cancer. Lung Cancer. 68:105–110.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Coulie PG, Karanikas V, Lurquin C, Colau
D, Connerotte T, Hanagiri T, Van Pel A, Lucas S, Godelaine D,
Lonchay C, et al: Cytolytic T-cell responses of cancer patients
vaccinated with a MAGE antigen. Immunol Rev. 188:33–42. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baba T, Shiota H, Kuroda K, Shigematsu Y,
Ichiki Y, Uramoto H, Hanagiri T and Tanaka F: Clinical significance
of human leukocyte antigen loss and melanoma-associated antigen 4
expression in smokers of non-small cell lung cancer patients. Int J
Clin Oncol. 18:997–1004. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dhodapkar MV, Osman K, Teruya-Feldstein J,
Filippa D, Hedvat CV, Iversen K, Kolb D, Geller MD, Hassoun H,
Kewalramani T, et al: Expression of cancer/testis (CT) antigens
MAGE-A1, MAGE-A3, MAGE-A4, CT-7 and NY-ESO-1 in malignant
gammopathies is heterogeneous and correlates with site, stage and
risk status of disease. Cancer Immun. 3:92003.PubMed/NCBI
|
|
6
|
Bolli M, Kocher T, Adamina M, Guller U,
Dalquen P, Haas P, Mirlacher M, Gambazzi F, Harder F, Heberer M, et
al: Tissue microarray evaluation of melanoma antigen E (MAGE)
tumor-associated antigen expression: Potential indications for
specific immunotherapy and prognostic relevance in squamous cell
lung carcinoma. Ann Surg. 236:785–793. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yakirevich E, Sabo E, Lavie O, Mazareb S,
Spagnoli GC and Resnick MB: Expression of the MAGE-A4 and NY-ESO-1
cancer-testis antigens in serous ovarian neoplasms. Clin Cancer
Res. 9:6453–6460. 2003.PubMed/NCBI
|
|
8
|
Yoshida N, Abe H, Ohkuri T, Wakita D, Sato
M, Noguchi D, Miyamoto M, Morikawa T, Kondo S, Ikeda H, et al:
Expression of the MAGE-A4 and NY-ESO-1 cancer-testis antigens and T
cell infiltration in non-small cell lung carcinoma and their
prognostic significance. Int J Oncol. 28:1089–1098. 2006.PubMed/NCBI
|
|
9
|
Zou W and Chen L: Inhibitory B7-family
molecules in the tumour microenvironment. Nat Rev Immunol.
8:467–477. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ostrand-Rosenberg S and Sinha P:
Myeloid-derived suppressor cells: Linking inflammation and cancer.
J Immunol. 182:4499–4506. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gabrilovich DI and Nagaraj S:
Myeloid-derived suppressor cells as regulators of the immune
system. Nat Rev Immunol. 9:162–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gabrilovich DI, Ostrand-Rosenberg S and
Bronte V: Coordinated regulation of myeloid cells by tumours. Nat
Rev Immunol. 12:253–268. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marcar L, Maclaine NJ, Hupp TR and Meek
DW: Mage-A cancer/testis antigens inhibit p53 function by blocking
its interaction with chromatin. Cancer Res. 70:10362–10370. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Srivastava MK, Zhu L, Harris-White M,
Huang M, St John M, Lee JM, Salgia R, Cameron RB, Strieter R,
Dubinett S, et al: Targeting myeloid-derived suppressor cells
augments antitumor activity against lung cancer. Immunotargets
Ther. 2012:7–12. 2012.PubMed/NCBI
|
|
15
|
Srivastava MK, Zhu L, Harris-White M, Kar
UK, Huang M, Johnson MF, Lee JM, Elashoff D, Strieter R, Dubinett
S, Sharma S, et al: Myeloid suppressor cell depletion augments
antitumor activity in lung cancer. PLoS One. 7:e406772012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Groeper C, Gambazzi F, Zajac P, Bubendorf
L, Adamina M, Rosenthal R, Zerkowski HR, Heberer M and Spagnoli GC:
Cancer/testis antigen expression and specific cytotoxic T
lymphocyte responses in non small cell lung cancer. Int J Cancer.
120:337–343. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cui NP, Xie SJ, Han JS, Ma ZF, Chen BP and
Cai JH: Effective adoptive transfer of haploidentical
tumor-specific T cells in B16-melanoma bearing mice. Chin Med J
(Engl). 125:794–800. 2012.PubMed/NCBI
|
|
18
|
Morales JK, Kmieciak M, Knutson KL, Bear
HD and Manjili MH: GM-CSF is one of the main breast tumor-derived
soluble factors involved in the differentiation of CD11b-Gr1- bone
marrow progenitor cells into myeloid-derived suppressor cells.
Breast Cancer Res Treat. 123:39–49. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baumgartner CK, Ferrante A, Nagaoka M,
Gorski J and Malherbe LP: Peptide-MHC class II complex stability
governs CD4 T cell clonal selection. J Immunol. 84:573–581. 2010.
View Article : Google Scholar
|
|
20
|
Wolchok JD, Hoos A, O'Day S, Weber JS,
Hamid O, Lebbé C, Maio M, Binder M, Bohnsack O, Nichol G, et al:
Guidelines for the evaluation of immune therapy activity in solid
tumors: Immune-related response criteria. Clin Cancer Res.
15:7412–7420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Daudi S, Eng KH, Mhawech-Fauceglia P,
Morrison C, Miliotto A, Beck A, Matsuzaki J, Tsuji T, Groman A and
Gnjatic S: Expression and immune responses to MAGE antigens predict
survival in epithelial ovarian cancer. PLoS One. 9:e1040992014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chaux P, Luiten R, Demotte N, Vantomme V,
Stroobant V, Traversari C, Russo V, Schultz E, Cornelis GR, Boon T,
et al: Identification of five MAGE-A1 epitopes recognized by
cytolytic T lymphocytes obtained by in vitro stimulation
with dendritic cells transduced with MAGE-A1. J Immunol.
163:2928–2936. 1999.PubMed/NCBI
|
|
23
|
Gunda V, Cogdill AP, Bernasconi MJ, Wargo
JA and Parangi S: Potential role of 5-aza-2′-deoxycytidine induced
MAGE-A4 expression in immunotherapy for anaplastic thyroid cancer.
Surgery. 154:1456–1462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ortiz ML, Lu L, Ramachandran I and
Gabrilovich DI: Myeloid-derived suppressor cells in the development
of lung cancer. Cancer Immunol Res. 2:50–58. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nagorsen D, Scheibenbogen C, Marincola FM,
Letsch A and Keilholz U: Natural T cell immunity against cancer.
Clin Cancer Res. 9:4296–4303. 2003.PubMed/NCBI
|
|
26
|
Cuenca AG, Cuenca AL, Winfield RD, Joiner
DN, Gentile L, Delano MJ, Kelly-Scumpia KM, Scumpia PO, Matheny MK,
Scarpace PJ, et al: Novel role for tumor-induced expansion of
myeloid-derived cells in cancer cachexia. J Immunol. 192:6111–6119.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duffour MT, Chaux P, Lurquin C, Cornelis
G, Boon T and van der Bruggen P: A MAGE-A4 peptide presented by
HLA-A2 is recognized by cytolytic T lymphocytes. Eur J Immunol.
29:3329–3337. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kobayashi T, Lonchay C, Colau D, Demotte
N, Boon T and van der Bruggen P: New MAGE-4 antigenic peptide
recognized by cytolytic T lymphocytes on HLA-A1 tumor cells. Tissue
Antigens. 62:426–432. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Y, Stroobant V, Russo V, Boon T and
van der Bruggen P: A MAGE-A4 peptide presented by HLA-B37 is
recognized on human tumors by cytolytic T lymphocytes. Tissue
Antigens. 60:365–371. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Scanlan MJ, Altorki NK, Gure AO,
Williamson B, Jungbluth A, Chen YT and Old LJ: Expression of
cancer-testis antigens in lung cancer: Definition of bromodomain
testis-specific gene (BRDT) as a new CT gene, CT9. Cancer Lett.
150:155–164. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schumacher K, Haensch W, Röefzaad C and
Schlag PM: Prognostic significance of activated CD8 (+) T cell
infiltrations within esophageal carcinomas. Cancer Res.
61:3932–3936. 2001.PubMed/NCBI
|
|
32
|
Cruz CR, Gerdemann U, Leen AM, Shafer JA,
Ku S, Tzou B, Horton TM, Sheehan A, Copeland A, Younes A, et al:
Improving T-cell therapy for relapsed EBV-negative Hodgkin lymphoma
by targeting upregulated MAGE-A4. Clin Cancer Res. 17:7058–7066.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Segura E and Villadangos JA: Antigen
presentation by dendritic cells in vivo. Curr Opin Immunol.
21:105–110. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Willimsky G and Blankenstein T: Sporadic
immunogenic tumours avoid destruction by inducing T-cell tolerance.
Nature. 437:141–146. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Drake CG, Jaffee E and Pardoll DM:
Mechanisms of immune evasion by tumors. Adv Immunol. 90:51–81.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Diaz-Montero CM, Salem ML, Nishimura MI,
Garrett-Mayer E, Cole DJ and Montero AJ: Increased circulating
myeloid-derived suppressor cells correlate with clinical cancer
stage, metastatic tumor burden and doxorubicin-cyclophosphamide
chemotherapy. Cancer Immunol Immunother. 58:49–59. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Meyer C, Cagnon L, Costa-Nunes CM,
Baumgaertner P, Montandon N, Leyvraz L, Michielin O, Romano E and
Speiser DE: Frequencies of circulating MDSC correlate with clinical
outcome of melanoma patients treated with ipilimumab. Cancer
Immunol Immunother. 63:247–257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shen P, Wang A, He M, Wang Q and Zheng S:
Increased circulating Lin(−/low) CD33(+) HLA-DR(−) myeloid-derived
suppressor cells in hepatocellular carcinoma patients. Hepatol Res.
44:639–650. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bianchi G, Borgonovo G, Pistoia V and
Raffaghello L: Immunosuppressive cells and tumour microenvironment:
Focus on mesenchymal stem cells and myeloid derived suppressor
cells. Histol Histopathol. 26:941–951. 2011.PubMed/NCBI
|
|
40
|
Fujimura T, Mahnke K and Enk AH: Myeloid
derived suppressor cells and their role in tolerance induction in
cancer. J Dermatol Sci. 59:1–6. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sevko A and Umansky V: Myeloid-derived
suppressor cells interact with tumors in terms of myelopoiesis,
tumorigenesis and immunosuppression: Thick as thieves. J Cancer.
4:3–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Umansky V and Sevko A: Tumor
microenvironment and myeloid-derived suppressor cells. Cancer
Microenviron. 6:169–177. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jungbluth AA, Ely S, DiLiberto M,
Niesvizky R, Williamson B, Frosina D, Chen YT, Bhardwaj N,
Chen-Kiang S, Old LJ, et al: The cancer-testis antigens CT7
(MAGE-C1) and MAGE-A3/6 are commonly expressed in multiple myeloma
and correlate with plasma-cell proliferation. Blood. 106:167–174.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Forghanifard MM, Gholamin M, Farshchian M,
Moaven O, Memar B, Forghani MN, Dadkhah E, Naseh H, Moghbeli M,
Raeisossadati R, et al: Cancer-testis gene expression profiling in
esophageal squamous cell carcinoma: Identification of specific
tumor marker and potential targets for immunotherapy. Cancer Biol
Ther. 12:191–197. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nagao T, Higashitsuji H, Nonoguchi K,
Sakurai T, Dawson S, Mayer RJ, Itoh K and Fujita J: MAGE-A4
interacts with the liver oncoprotein gankyrin and suppresses its
tumorigenic activity. J Biol Chem. 278:10668–10674. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Park JH, Kong GH and Lee SW: hMAGE-A1
overexpression reduces TNF-alpha cytotoxicity in ME-180 cells. Mol
Cells. 14:122–129. 2002.PubMed/NCBI
|
|
47
|
Iclozan C, Antonia S, Chiappori A, Chen DT
and Gabrilovich D: Therapeutic regulation of myeloid-derived
suppressor cells and immune response to cancer vaccine in patients
with extensive stage small cell lung cancer. Cancer Immunol
Immunother. 62:909–918. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Waight JD, Netherby C, Hensen ML, Miller
A, Hu Q, Liu S, Bogner PN, Farren MR, Lee KP, Liu K, et al:
Myeloid-derived suppressor cell development is regulated by a
STAT/IRF-8 axis. J Clin Invest. 123:4464–4478. 2013. View Article : Google Scholar : PubMed/NCBI
|