Introduction
Lung cancer has one of the lowest survival rates of
all types of cancer, accounting for 1.59 million mortalities in the
world in 2012 (1). Smoking,
particularly of cigarettes, is estimated to cause 87% of the lung
cancer mortalities in men, and 70% of the lung cancer mortalities
in women (2). Cigarette smoke
contains numerous carcinogens, of which, polycyclic aromatic
hydrocarbons and the tobacco-specific nitrosamine,
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol, are likely to serve
major roles (3). However, recent
evidence suggests that cigarette smoking not only promotes lung
carcinogenesis, but that it also promotes the progression of lung
cancer (4). For example, continued
smoking during lung cancer treatment has been associated with
decreased survival (5), while smoking
cessation during the treatment improves the therapeutic outcome
(6). Furthermore, cigarette smoking
increases the risk of recurrence and the cancer-specific mortality
in non-cigarette smoke-associated cancers, including prostate
cancer (7,8). Nicotine is one of the components of
cigarette smoke that may promote cancer progression.
Whilst it does not exert direct carcinogenic
activity, nicotine is commonly accepted instead as a tumor promoter
(4). Recent evidence suggests that
the tumor-promoting effects of nicotine result in increased
proliferation, invasion, mesenchymal transition (EMT) and
angiogenesis, and decreased cell death and apoptosis (4,9–17). However, the molecular mechanisms by
which nicotine promotes tumor progression are not yet fully
understood.
Cell motility is a critical event during the
invasion and metastasis of cancer cells. During the present study,
the effect of nicotine on the motility of lung cancer A549 cells
was investigated. It was demonstrated that nicotine, by itself,
does not induce A549 cell migration, but instead promotes
hepatocyte growth factor (HGF)-induced migration. The
tumor-promoting effect of nicotine was mediated by its binding to
α7-nicotine acetylcholine receptors (α7-nAchR) and the subsequent
upregulation of the HGF-stimulated phosphoinositide kinase-3
(PI3K)/Akt signaling pathway. The results of the present study
indicate that nicotine enhances the pro-migratory effect of HGF on
lung cancer cells, thereby potentially contributing to lung cancer
progression.
Materials and methods
Cell culture
Lung cancer A549 cells were purchased from the
American Type Culture Collection (Manassas, VA, USA). The cells
were grown and maintained on bovine type I collagen-coated (Nippi,
Inc., Tokyo, Japan) plates in Dulbecco's modified Eagle's medium
(DMEM; Gibco®; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
containing 10% fetal bovine serum (FBS) at 37°C in a humidified
incubator saturated with a gas mixture containing 5%
CO2.
In vitro wound healing assay
The migratory activity of the cells was determined
by the in vitro wound healing assay (18). A549 cells were grown to confluence in
24-well plates. The cells were serum-starved in 0.5% FBS for 48 h
and were subsequently washed with phosphate-buffered saline (PBS).
The cells were then scratched in a straight line with a sterile
200-ml tip and washed twice with PBS. Following this, the cells
were incubated in serum-free DMEM containing or lacking HGF (20
ng/ml; PromoCell GmbH, Heidelberg, Germany) and in the presence or
absence of nicotine (0.1, 1 and 10 µM; Wako Pure Chemical
Industries, Ltd., Osaka, Japan). In several experiments, medium was
added with specific inhibitors 1 h prior to the treatment with HGF
and nicotine. The specific inhibitors included: Methyllycaconitine
(10 µM; Sigma-Aldrich Japan, Tokyo, Japan), an inhibitor of
α7-nAchR, LY294002 (5 µM; Cayman Chemical Company, Ann Arbor, MI,
USA), an inhibitor of PI3K, and PD98059 (5 µM; Cayman Chemical
Company), an inhibitor of mitogen-activated protein kinase
(MAPK)/extracellular signal related kinase (ERK) kinase (MEK).
Following 12 or 18 h, the cells were fixed with 10% formalin and
washed twice with PBS. Wound healing was observed with the Olympus
Model IX71 Fluorescence Microscope with phase contrast (Olympus
Corporation, Tokyo, Japan) equipped with a digital camera. The area
of the wound was measured using image analysis software (Win Roof
Version 3.5; Mitani Corporation, Fukui, Japan) on a Microsoft XP
computer. A total of 5 measurements were taken from 5 fields of
each well, obtained from 6 wells in each experiment.
Cell-based protein phosphorylation
assay
The A549 cells were grown to 90% confluence in
96-well plates and serum-starved in DMEM with 0.5% FBS. The cells
were subsequently treated with HGF (20 ng/ml) and/or nicotine (10
µM) for 2 h, and fixed with 10% formalin. Phosphorylation of ERK1/2
and Akt protein was assayed using the phospho-ERK1/2 antibody or
the phospho-Akt antibody from Fast Activated Cell-based ELISA kits
(Active Motif, Carlsbad, CA, USA), according to the manufacturer's
protocols. Absorbance was measured using a Benchmark Plus
microplate spectrophotometer (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) at 450 nm, with a reference wavelength of 655 nm.
Following this, the wells were stained with a crystal violet
solution and the absorbance at 595 nm was measured. The protein
phosphorylation levels were corrected for the cell number by
dividing the optical density (OD)450 reading for a given
well by the OD595 reading for that well.
Statistical analysis
The statistical analyses were performed using Excel
X software with the add-in software Statcel 3 (OMS Inc., Tokyo,
Japan). Data are expressed as the mean ± standard error of the
mean. Statistical differences were analyzed by analysis of
variance, and if the results were significant, the Tukey-Kramer
test was employed as a multiple comparison post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Nicotine promotes the migration of
HGF-treated A549 cells
The migration of cancer cells is a vital step in
cancer invasion and metastasis. To determine the effect of nicotine
on A549 cell migration, a wound healing assay was conducted, which
is typically considered to be a simple and reliable test for the
evaluation of cell motility. During the assay, the cells were
maintained in serum-free medium in order to avoid any potential
modulatory effect that the serum may have on cell motility. As
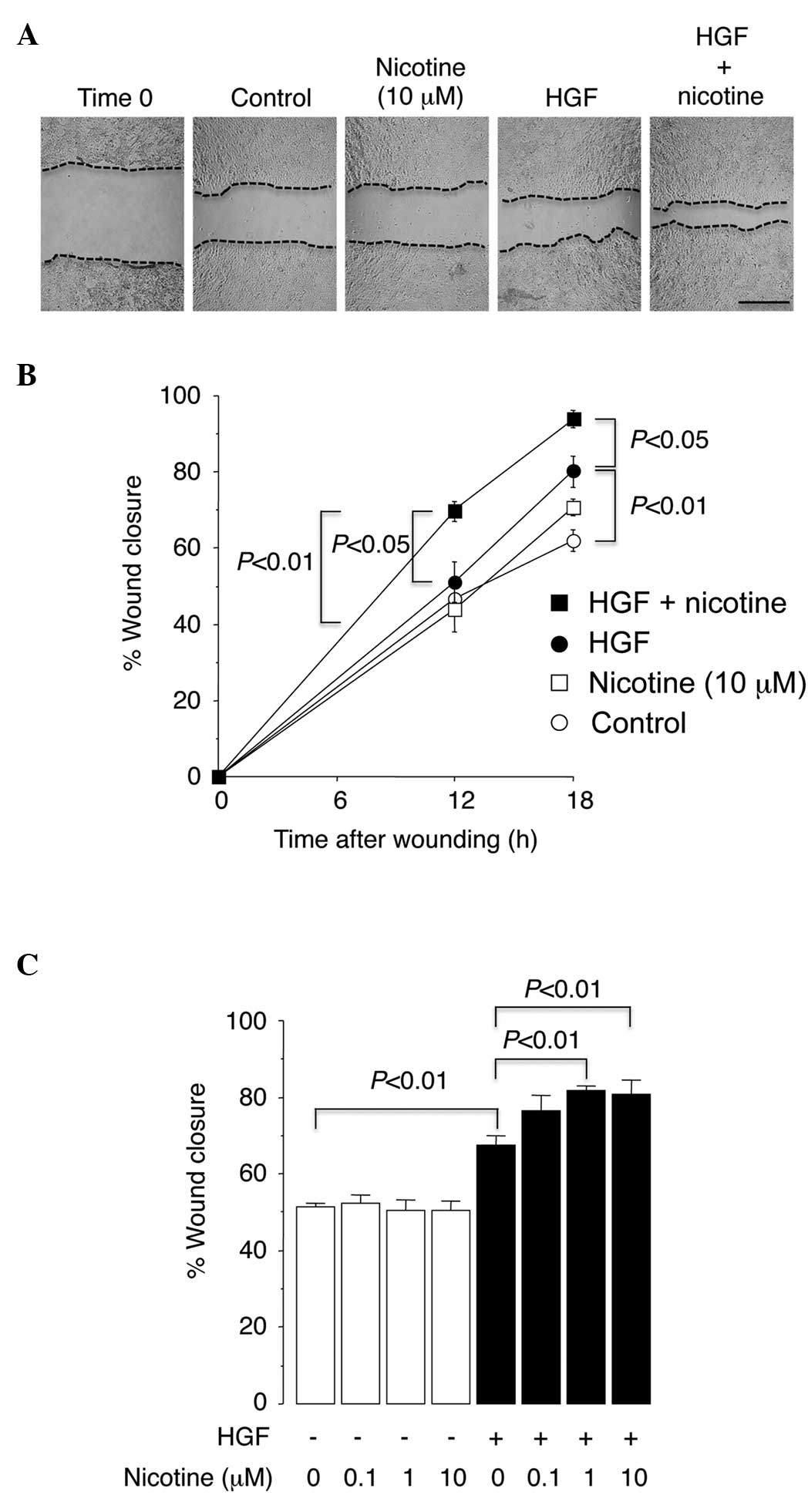
presented in Fig. 1, it was observed
that nicotine had no effect on cell migration towards the scratched
area in serum-free medium, suggesting that nicotine does not induce
cell migration when acting alone. Subsequently, it was examined
whether nicotine affects cell migration induced by a pro-migratory
stimulus. HGF, which has been demonstrated to be the major
pro-migratory stimulus for epithelial cells, including A549 cells,
was utilized for this purpose (19–21). As
expected, HGF treatment increased cell migration towards the
scratched area (Fig. 1). When 0.1 and
1 µM nicotine was added to the HGF-treated cells, the cell
migratory activity significantly increased (P<0.01; Fig. 1C). Such results suggest that nicotine
does not induce cell migration working alone, but that it instead
promotes the cell migratory response to the pro-migratory stimulus
HGF.
Nicotine promotes HGF-induced cell
migration through α7-nAchR
To determine whether the effect of nicotine in
promoting HGF-induced cell migration is mediated by activation of
the nicotine receptor, the A549 cells were pretreated with the
α7-nAchR antagonist, methyllycaconitine, prior to adding nicotine
to the HGF-treated cells. As presented in Fig. 2, pretreatment with methyllycaconitine
abrogated the effect of nicotine in promoting the cell migration
induced by HGF treatment (P<0.05). These results suggest that
nicotine enhances the pro-migratory effect of HGF on A549 cells
through the activation of α7-nAchR-mediated signaling pathways.
Nicotine promotes HGF-induced cell
migration by potentiating PI3K/Akt activation
HGF binds to and activates its only known receptor,
c-Met, a receptor tyrosine kinase that mediates downstream
signaling cascades (20–23). Results obtained thus far indicate that
the nicotine/α7-nAchR-mediated signaling pathway interacts with the
HGF/c-Met-mediated signaling pathway. The downstream signals of
HGF/c-Met include ERK1/2 and PI3K/Akt (20,21). The
current study therefore investigated the potential involvement of
these downstream signals in the mediation of HGF-induced cell
migration. It was demonstrated that pretreatment of the cells with
the PI3K inhibitor, LY294002, significantly inhibited the effect of
HGF in promoting cell migration (P<0.05; Fig. 3). Similar results were observed
following pretreatment of the cells with the MEK inhibitor,
PD98059, although the effect was not statistically significant
(Fig. 3). These results suggest that
the activation of PI3K/Akt is required for HGF/c-Met-mediated
downstream signaling events that result in A549 cell migration.
Effect of nicotine on the activation
of PI3K/Akt
As previously reported (21), HGF treatment induced the
phosphorylation of Akt in the A549 cells (P<0.05; Fig. 4A). Nicotine alone did not induce Akt
phosphorylation, but it was noted that it enhanced the HGF-induced
phosphorylation of Akt when compared with HGF alone (P<0.01).
While HGF treatment also significantly induced the phosphorylation
of ERK1/2 (P<0.05; Fig. 4B,
nicotine had no such effect on the phosphorylation level of ERK1/2
in either the HGF-treated or HGF-untreated cells. When combined,
these results indicate that nicotine enhances HGF-induced cell
migration through the activation of α7-nAchRs, whilst also
amplifying the HGF-induced activation of PI3K/Akt.
Discussion
During the present study, it was demonstrated that
nicotine does not induce A549 cell migration alone, but that it
does promote HGF-induced cell migration. Furthermore, it was also
observed that the promotion of HGF-induced cell migration by
nicotine mediated its binding to α7-nAchR, and that as a result,
the HGF-mediated PI3K/Akt signaling pathway is amplified. Such
results indicate that nicotine is not a pro-migratory factor by
itself, but that it instead enhances the cell-migratory
responsiveness to the pro-migratory stimulus of HGF.
The findings that nicotine did not induce migration
in quiescent A549 cancer cells in the absence of HGF is
inconsistent with the results of previous studies, which observed
that nicotine itself exerts a pro-migratory effect in various types
of cancer cells (10,12–14,16,17).
The reasoning behind the inconsistent results between the present
study and previous studies remains uncertain, however, it may be
attributed to differences in the experimental protocol. The current
study performed a wound healing assay in serum-free medium
following 48 h of serum starvation, while in previous studies, the
assay was performed in serum-supplemented medium (10,12,14,17),
and/or without serum starvation prior to inflicting the scratch
(16). Since serum may contain
pro-migratory factors, including HGF, serum contamination during
the wound healing assay may therefore potentiate cell migration
following the addition of nicotine.
HGF serves a critical role in cancer progression by
upregulating cell proliferation, migration, invasion and
angiogenesis (22). Elevated HGF and
c-Met expression, as well as numerous c-Met mutations, have been
reported in human cancer, including lung cancer, and have been
demonstrated to be correlated with a poor prognosis (22). Accordingly, c-Met has been proposed as
a possible therapeutic target for the treatment of lung cancer
(23). However, the
HGF/c-Met-mediated signaling pathway is complex. Recent evidence
suggests that c-Met cross-talks with other receptors, including the
epidermal growth factor receptor and the recepteur d'origine
nantais (23). To the best of our
knowledge, the present study provides evidence of crosstalk between
HGF/c-Met and nicotine/nAchR for the first time.
The current study demonstrated that the PI3K
inhibitor, LY294002, inhibited cell migration that had been induced
by HGF treatment, corroborating the results of previous studies and
establishing that PI3K/Akt is an essential mediator downstream of
HGF/c-Met (20,22,23). It
was also demonstrated that Akt was activated by HGF, but not by
nicotine. However, Akt activation by HGF treatment was enhanced
when the cells were co-treated with nicotine. These findings
suggest that nicotine upregulates the HGF-induced activation of the
PI3K/Akt pathway, leading to the enhancement of the cell migratory
response to HGF.
nACHRs mediate a number of the effects exerted by
nicotine (24). Among them, α7-nAchR
has been demonstrated to be expressed in cancer cells, including
lung cancer A549 cells (24,25). Lending support to the demonstration of
cross-talk between nicotine/α7-nAchR and HGF/c-Met in the present
study, previous studies have established that nicotine/α7-nAchR
cross-talks with the downstream signaling of various growth
factors, including epithelial cell growth factor, basic fibroblast
growth factor and vascular endothelial cell growth factor,
promoting cancer cell proliferation (4,17,24). However, the mechanisms underlying
signal crosstalk are complex. The binding of nicotine to nAchR
increases Ca2+ influx, thereby potentially modulating
Ca2+-dependent signal transduction (11,24).
Nicotine may also increase the expression of receptor tyrosine
kinases and the recruitment of growth/migration-associated factors
to the surface of cells (17,24). The effects of nicotine demonstrated in
the present study are unlikely to be due to direct activation of
PI3K/Akt by nicotine, as nicotine alone did not induce
phosphorylation of Akt in the serum-free condition in the absence
of HGF. It is conceivable that nicotine/α7-nAchR may act in synergy
with the HGF/c-Met-induced pathway, elevating the sensitivity of
cancer cells to HGF, and thereby promoting cell migration.
In the present study, nicotine enhanced HGF-induced
cell migration at concentrations of 1 and 10 µM. This range of
nicotine concentrations reasonably mimics the nicotine
concentrations observed in vivo in humans. While plasma
concentrations of nicotine in chronic smokers are reported to range
between 0.01 and 1 µM (26), lung
tissue concentrations of nicotine may be much higher, due to the
direct contact of the lung tissue with nicotine in cigarette
smoke.
In conclusion, the current study demonstrated that
nicotine treatment potentiates the migratory responsiveness of lung
cancer cells to HGF. Cumulative evidence suggests that nicotine may
have a broad spectrum of tumor-promoting activities, which include
increasing cell proliferation, invasion, EMT and angiogenesis
(4). The present study provides an
insight into the mechanism of the promotion of tumor cell
activities initiated by nicotine, by demonstrating how
nicotine-modulated growth factor-mediated signal transduction leads
to increased cell motility, a vital step in cancer invasion and
metastasis. Furthermore, the results also provide evidence of the
inhibition of α7-nAchR as a potential therapeutic target.
References
|
1
|
World Health Organization: World Cancer
Report. 2014.https://www.iarc.fr/en/publications/books/wcr/wcr-order.phpAccessed.
October 30–2015
|
|
2
|
American Cancer Society. Cancer facts and
figures. 2014.http://www.cancer.org/acs/groups/content/@research/documents/webcontent/acspc-042151.pdfAccessed.
October 30–2015
|
|
3
|
Hecht SS: Lung carcinogenesis by tobacco
smoke. Int J Cancer. 131:2724–2732. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Warren GW and Singh AK: Nicotine and lung
cancer. J Carcinog. 12:12013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Videtic GM, Stitt LW, Dar AR, Kocha WI,
Tomiak AT, Truong PT, Vincent MD and Yu EW: Continued cigarette
smoking by patients receiving concurrent chemoradiotherapy for
limited-stage small-cell lung cancer is associated with decreased
survival. J Clin Oncol. 21:1544–1549. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andreas S, Rittmeyer A, Hinterthaner M and
Huber RM: Smoking cessation in lung cancer-achievable and
effective. Dtsch Arztebl Int. 110:719–724. 2013.PubMed/NCBI
|
|
7
|
Joshu CE, Mondul AM, Meinhold CL,
Humphreys EB, Han M, Walsh PC and Platz EA: Cigarette smoking and
prostate cancer recurrence after prostatectomy. J Natl Cancer Inst.
103:835–838. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kenfield SA, Stampfer MJ, Chan JM and
Giovannucci E: Smoking and prostate cancer survival and recurrence.
JAMA. 305:2548–2555. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warren GW, Romano MA, Kudrimoti MR,
Randall ME, McGarry RC, Singh AK and Rangnekar VM: Nicotinic
modulation of therapeutic response in vitro and in vivo. Int J
Cancer. 131:2519–2527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu L and Deng X: Protein kinase Ciota
promotes nicotine-induced migration and invasion of cancer cells
via phosphorylation of micro- and m-calpains. J Biol Chem.
281:4457–4466. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Q, Tang X, Zhang ZF, Velikina R, Shi
S and Le AD: Nicotine induces hypoxia-inducible factor-1alpha
expression in human lung cancer cells via nicotinic acetylcholine
receptor-mediated signaling pathways. Clin Cancer Res.
13:4686–4694. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo J, Ibaragi S, Zhu T, Luo LY, Hu GF,
Huppi PS and Chen CY: Nicotine promotes mammary tumor migration via
a signaling cascade involving protein kinase C and CDC42. Cancer
Res. 68:8473–8481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dasgupta P, Rizwani W, Pillai S, Kinkade
R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, et
al: Nicotine induces cell proliferation, invasion and
epithelial-mesenchymal transition in a variety of human cancer cell
lines. Int J Cancer. 124:36–45. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lien YC, Wang W, Kuo LJ, Liu JJ, Wei PL,
Ho YS, Ting WC, Wu CH and Chang YJ: Nicotine promotes cell
migration through alpha7 nicotinic acetylcholine receptor in
gastric cancer cells. Ann Surg Oncol. 18:2671–2679. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi D, Guo W, Chen W, Fu L, Wang J, Tian
Y, Xiao X, Kang T, Huang W and Deng W: Nicotine promotes
proliferation of human nasopharyngeal carcinoma cells by regulating
α7AChR, ERK, HIF-1α and VEGF/PEDF signaling. PLoS One.
7:e438982012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Momi N, Ponnusamy MP, Kaur S, Rachagani S,
Kunigal SS, Chellappan S, Ouellette MM and Batra SK:
Nicotine/cigarette smoke promotes metastasis of pancreatic cancer
through α7nAChR-mediated MUC4 upregulation. Oncogene. 32:1384–1395.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Khalil AA, Jameson MJ, Broaddus WC, Lin PS
and Chung TD: Nicotine enhances proliferation, migration, and
radioresistance of human malignant glioma cells through EGFR
activation. Brain Tumor Pathol. 30:73–83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsuji T, Aoshiba K, Itoh M, Nakamura H and
Yamaguchi K: Hypercapnia accelerates wound healing in endothelial
cell monolayers exposed to hypoxia. Open Respir Med J. 7:6–12.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stoker M, Gherardi E, Perryman M and Gray
J: Scatter factor is a fibroblast-derived modulator of epithelial
cell mobility. Nature. 327:239–242. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ponzetto C, Bardelli A, Zhen Z, Maina F,
dalla Zonca P, Giordano S, Graziani A, Panayotou G and Comoglio PM:
A multifunctional docking site mediates signaling and
transformation by the hepatocyte growth factor/scatter factor
receptor family. Cell. 77:261–271. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Forte G, Minieri M, Cossa P, Antenucci D,
Sala M, Gnocchi V, Fiaccavento R, Carotenuto F, De Vito P, Baldini
PM, et al: Hepatocyte growth factor effects on mesenchymal stem
cells: Proliferation, migration, and differentiation. Stem Cells.
24:23–33. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maroun CR and Rowlands T: The Met receptor
tyrosine kinase: A key player in oncogenesis and drug resistance.
Pharmacol Ther. 142:316–338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sadiq AA and Salgia R: MET as a possible
target for non-small-cell lung cancer. J Clin Oncol. 31:1089–1096.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Niu XM and Lu S: Acetylcholine receptor
pathway in lung cancer: New twists to an old story. World J Clin
Oncol. 5:667–676. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dasgupta P, Kinkade R, Joshi B, Decook C,
Haura E and Chellappan S: Nicotine inhibits apoptosis induced by
chemotherapeutic drugs by up-regulating XIAP and survivin. Proc
Natl Acad Sci USA. 103:6332–6337. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Benowitz NL: Drug therapy. Pharmacologic
aspects of cigarette smoking and nicotine addiction. N Engl J Med.
319:1318–1330. 1988. View Article : Google Scholar : PubMed/NCBI
|