Introduction
Molecular targeted therapies have become the most
promising potential treatment for lung cancer, the leading cause of
cancer-associated mortality worldwide (1). As a generally accepted treatment target,
epidermal growth factor receptor (EGFR) is involved in numerous
carcinogenic processes, including cell invasion, proliferation,
apoptosis and angiogenesis (2). Two
small molecule inhibitors of EGFR (erlotinib and gefitinib) have
been used extensively in clinical settings and have demonstrated
marked effects in the treatment of patients with non-small cell
lung cancer (NSCLC) with activating EGFR mutations (3,4). However,
a number of patients do not benefit from such inhibitors, and even
those who initially exhibit a positive response to EGFR inhibitors
may eventually become refractory (5).
It has been reported that the remission rate of gefitinib and
erlontinib used in the second- or third-line of NSCLC treatment is
9–27%, and among these cases 50% relapse 6–12 months later
(5). To further improve the efficacy
of EGFR tyrosine kinase inhibitors (TKIs), a greater understanding
of the mechanisms underlying such resistance is required.
To date, the mechanisms underlying resistance to
EGFR TKIs have mainly focused on several aspects: Secondary
mutation of the EGFR gene exon 20 (6), the constitutive activation of downstream
signaling pathways, epithelial-mesenchymal transition (EMT)
associated with intrinsic and acquired resistance (7,8), c-MET
gene amplification (9), increased
angiogenesis and tumor micro-circumstance associated drug
resistance (7,10). Recently, cell adhesion-mediated drug
resistance (CAM-DR) has attracted increasing attention. Changes in
the tumor microenvironment, including adhesion molecules and their
receptors, cytokines and the extracellular matrix (ECM), for
example fibronectin and collagen I, may induce resistance of NSCLC
to EGFR TKIs (10). Integrin is one
of the most significant adhesion molecules that has been suggested
to be involved in CAM-DR (11).
Integrins are transmembrane heterodimers that consist of
non-covalently bound α and β glycoprotein subunits and transmit
biomechanical cues to mediate cell-ECM interactions (12,13). The
loss of integrin-mediated anchorage to the ECM may result in cell
apoptosis, known as anoikis, in epithelial or endothelial cells,
indicating a significant role for this molecule in the control of
tumorigenesis. It has also been reported that integrins mediating
cell-ECM signaling are able to affect various tumor cell behaviors,
including proliferation, survival, invasion and metastasis
(14,15). Increasing evidence has suggested that
the integrin β1 signaling pathway has a significant role in
mediating resistance to chemotherapies by enhancing cell survival
in myeloma, glioblastoma, ovarian cancer and lung cancer (16–19). Given
that integrins and EGFR share a number of downstream signaling
pathways, including protein kinase B (Akt) (20), it was hypothesized that integrin may
be involved in inducing the resistance of NSCLC to EGFR TKIs.
In the present study, a gefitinib-resistant PC9/G
cell line was generated by exposure of PC9 cells to mutagen,
N-methyl-N'-nitro-N-nitrosoguanidine (MNNG), with
gefitinib, and it was found that expression of integrin β1 was
higher in PC9/G cells than that in the parental cell line, PC9.
Knockdown of integrin β1 by RNA interference (RNAi) was able to
restore the sensitivity of PC9/G cells to gefitinib. Furthermore,
it was demonstrated that such a restoration may be induced via
inactivation of the phosphoinositide 3-kinase (PI3K) pathway. These
results therefore reveal a potential mechanism by which NSCLC may
acquire resistance to EGFR antagonists.
Materials and methods
Reagents
Gefitinib was purchased from AstraZeneca
(Macclesfield, UK). The PI3K inhibitor, LY294002, was purchased
from Sigma-Aldrich (St. Louis, MO, USA). Monoclonal human
anti-mouse integrin β1 antibody (1:1,000; cat. no. CBL481P) was
obtained from Chemicon® (Merck Millipore, Darmstadt, Germany).
Monoclonal human anti-rabbit c-MET (1:1,000; cat. no. 8198),
polyclonal human anti-rabbit Akt (1:1,000; cat. no. 9272),
polyclonal human anti-rabbit phosphorylated (phospho)-Akt (Ser473;
1:1,000; cat. no. 9271), polyclonal human anti-rabbit Erk (1:1,000;
cat. no. 4695), monoclonal human anti-rabbit phospho-Erk (1:1,000;
cat. no. 4370), polyclonal human anti-rabbit EGFR (1:1,000; cat.
no. 2232) and polyclonal human anti-rabbit phospho-EGFR (1:1,000;
cat. no. 2234) antibodies were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Monoclonal rabbit anti-mouse
GAPDH antibody (1:10,000; cat. no. KC-5G5) was purchased from
Shanghai Kangchen Bio-technology Co. (Shanghai, China). Gibco
Dulbecco's modified Eagle's medium was from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA).
Cell culture and transfection
The PC9 human lung adenocarcinoma cell line (passage
number, 10; negative for mycoplasmic infection) was provided by Dr
Takayama (Kyushu University, Fukuoka, Japan). A gefitinib-resistant
PC9/G cell line was established according to the protocol reported
by Koizumi et al (21).
Briefly, cultured PC9 cells were exposed to 2.5 µg/ml MNNG
(Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) for 24 h,
washed and cultured in medium supplemented with 0.2 µM gefitinib
for 7 days. Following exposure to gefitinib, cells were washed and
cultured in drug-free medium (Gibco; Thermo Fisher Scientific,
Inc.) for 14 days. When the number of variable cells had increased
to 90%, as determined by trypan blue (Sigma-Aldrich) exclusion,
cells (3×103/ml) were seeded on 96-well culture plates
in medium containing 0.3–0.5 µM gefitinib for subcloning. Following
21 days of culture, colonies were harvested by trypsinization and a
single clone was obtained. The cell line was maintained in medium
containing 0.05 µmol/l gefitinib at 37°C in a humidifed atmosphere
of 5% CO2.
Integrin β1 short hairpin RNA (shRNA)-pRNAT-U6.1/Neo
vectors were generated by Shanghai Kangchen Bio-technology Co. To
construct integrin β1 shRNA, sense and antisense DNA
oligonucleotides were designed from double-stranded RNA with a loop
structure: Integrin β1 shRNA [short interfering (si)-ITGB1] sense,
5′-GGATTCTGACAGCTTTAAA-3′ and antisense, 5′-TTTAAAGCTGTCAGAATCC-3′.
A scrambled sequence (si-scrambled; 5′-TTCTCCGAACGTGTACGT-3′;
Shanghai Kangchen Bio-technology Co.) was used as a control. Cells
were transfected with Lipofectamine® 2000 reagent (Invitrogen;
Thermo Fisher Scientific) according to the manufacturer's protocol.
The transient transfection efficiency for PC9 and PC9/G cells was
65–75%, as determined by green fluorescent protein plasmid
transfection. Briefly, PC9 and PC9/G cells were grown to 80%
confluence on six-well plates, washed twice with serum free medium
(Gibco; Thermo Fisher Scientific, Inc.), resuspended in
antibiotic-free DMEM (5×105 cells/ml; Gibco; Thermo
Fisher Scientific, Inc.) and transfected with 16 µg/ml total DNA
using Lipofectamine 2000 (1:5 ratio). After 6 h, the transfection
medium was removed and medium containing 2% fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.) was added to induce
transgene expression. Following transfection for 24 h, 48 h, 72 h,
4 and 5 days, respectively, the number of cells exhibiting green
GFP fluorescence and the total number of cells was counted using a
fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Transfection efficiency (%) was calculated using the following
formula: (number of positive cells/number of total cells) ×
100.
Gene status and gene expression
analysis
In order to evaluate EGFR sequence variations,
polymerase chain reaction (PCR) amplifications of genomic DNA
isolated from NSCLC cell lines were conducted using nested primers
to amplify EGFR exons 18–21. Briefly, total DNA was extracted from
cells using DNA extraction kits [Tiangen Biotech (Beijing) Co.,
Ltd., Beijing, China]. The following primers were used: Forward,
5′-CAAATGAGCTGGCAAGTGCCGTGTC-3′ and reverse,
5′-GAGTTTCCCAAACACTCAGTGAAAC-3′ for external exon 18; Forward,
5′-CAAGTGCCGTGTCCTGGCACCCAA GC-3′ and reverse,
5′-CCAAACACTCAGTGAAACAAAGAG-3′ for internal exon 18; forward,
5′-GCAATATCAGCCTTAGGTGCGGCTC-3′ and reverse,
5′-CATAGAAAGTGAACATTTAGGATGTG-3′ for external exon 19; forward,
5′-CCTTAGGTGCGGCTCCACAGC-3′ and reverse,
5′-CATTTAGGATGTGGAGATGAGC-3′ for internal exon 19; forward,
5′-CCATGAGTACGTATTTTGAAACTC-3′ and reverse,
5′-CATATCCCCATGGCAAACTCTTGC-3′ for external exon 20; forward,
5′-GAAACTCAAGATCGCATTCATGC-3′ reverse,
5′-GCAAACTCTTGCTATCCCAGGAG-3′ for internl exon 20; forward,
5′-CTAACGTTCGCCAGCCATAAGTCC-3′ and reverse,
5′-GCTGCGAGCTCACCCAGAATGTCTGG-3′ for external exon 21; forward,
5′-CAGCCATAAGTCCTCGACGTGG-3′ and reverse,
5′-CATCCTCCCCTGCATGTGTTAAAC-3′ for internal exon 21. A total of 50
ng total DNA was used as a template for each reaction. PCR was
performed under the following conditions: Pre-incubation at 95°C
for 15 min, followed by amplification for 35 cycles (95°C for 20
sec, 60°C for 30 sec, 72°C for 1 min) with a final extension step
at 72°C for 10 min. Subsequently, the products were directly
sequenced by Invitrogen (Thermo Fisher Scientific, Inc.). Basal
gene expression analysis of NSCLC cell lines was performed on RNA
extracted from subconfluent cell cultures, using the BiostarH-140s
microarray platform (China United Gene Health Industry Ltd., Hong
Kong, China). Briefly, total RNA was extracted using the Trizol
Plus Kit (Takara Bio, Inc., Shiga, Japan). A fluorescent labeling
cDNA probe was generated from 3 µg RNA using reverse transcriptase,
Cy3-dCTP and Cy5-dCTP (all purchased from Shanghai BioStar Genechip
Inc., Shanghai, China). Labeled cDNA was then hybridized using the
BiostarH-140s cDNA microarray (Shanghai Biostar Genechip Inc.) at
42°C. The array was imaged using the ScanArray 4000 scanner (GSI
Group, Inc., Bedford, MA, USA) and images were processed by ImaGene
3.0 (GSI Group, Inc.).
Cell proliferation assay
The inhibition of cell proliferation was evaluated
using the tetrazolium dye [3-(4,5-dimethylthiazol-2-yl)
2,5-diphenyltetrazolium bromide (MTT); Sigma-Aldrich] assay. Cells
were seeded into 96-well plates at a density of 10,000 cells/well.
Twenty-four hours following seeding, cells were exposed to various
concentrations of gefitinib (0, 0.001, 0.005, 0.01, 0.05, 5.0,
10.0, 20.0 and 40.0 µmol/l) with or without LY294002 (25 µmol/l).
Seventy-two hours following drug treatment, cells were incubated
with MTT (5 mg/ml) for 4 h at 37°C. Culture medium containing MTT
was subsequently removed and formazan crystals were dissolved in
200 µl dimethyl sulfoxide (Shanghai Pharmaceuticals Holding Co.,
Ltd., Shanghai, China). Cell proliferation inhibition was
determined by measuring the absorbance at 530 nm using a Multiskan
MK3 microplate reader (Thermo Labsystems, Helsinki, Finland). These
experiments were performed in quadruplicate on three separate
occasions.
Cell cycle analysis
Cells (4×105 cells/ml) in the logarithmic
growth phase were seeded into six-well plates. The two cell lines
(PC9 and PC9/G cells) were incubated for 24 h in serum free medium
(Gibco; Thermo Fisher Scientific, Inc.) to arrest the cell cycle in
the G0 phase. The cells were then incubated in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.). After ~48 h
incubation, the cell lines were treated with 0.03 µmol/l gefitinib,
respectively. Cells were harvested by trypsinization, fixed in 70%
ice-cold ethanol for 2 h and stained with propidium iodide (Beijing
Biosea Biotechnology Co. Ltd., Beijing, China). Cell cycle analysis
was performed using a FC500 flow cytometer (Beckman Coulter, Inc.,
Brea, CA, USA).
PCR
Integrin β1 RNA was amplified from the complementary
DNA of PC9 and PC9/G by the SYBR Green real-time PCR kit (Toyobo
Co., Ltd., Osaka, Japan). Integrin β1 forward, CAAAGGAACAGCAGAGAAGC
and reverse, ATTGAGTAAGACAGGTCCATAAGG. GAPDH forward,
TGGTATCGTGGAAGGACTCATGAC and reverse, ATGCCAGTGAGCTTCCCGTTCAGC.
Following 3 min of pre-incubation at 95°C, PCR amplification was
performed for 40 cycles (94°C for 5 sec, 61°C for 34 sec), followed
by a final melting curve step (from 60 to 95°C; 1°C/min). The
2−ΔΔCq method was used to analyze the relative
quantitative expression levels of integrin β1, while GAPDH
functioned as an internal control gene.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay
buffer (Beyotime Institute of Biotechnology, Shanghai, China). The
Bradford assay (22) was used to
determine total protein concentrations, which were normalized to 1
µg/µl for all samples. Subsequently, samples were prepared in
loading buffer (Beyotime Institute of Biotechnology) and heated to
95°C for 10 min. The samples were separated on 8% polyacrylamide
gels for total EGFR, phospho-EGFR and integrin β1, and 10% gels for
total Akt, phospho-Akt, c-MET, Erk and phospho-Erk. Briefly,
protein lysates (20 µl) in loading buffer from each cell line were
loaded into each well. Wet transfer was performed for 2.5 h at
constant current (200 mA) using nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA). The membrane was subsequently
blocked for 1 h in 5% non-fat milk in 0.2% Tris-buffered saline
Tween 20 (TBST; Beyotime Institute of Biotechnology). The membrane
was then washed three times in 0.2% TBST for 10 min each and
incubated overnight at 4°C with primary antibodies against total
EGFR, phospho-EGFR, integrin β1, total Akt, phospho-Akt, c-MET, Erk
or phospho-Erk, respectively. Subsequently, the membrane was washed
three times in 0.2% TBST for 10 min each. Horseradish
peroxidase-conjugated goat anti-rabbit (cat. no. sc-2004) or
anti-mouse (cat. no. sc-2055) immunoglobulin G (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) were used as secondary
antibodies for enhanced chemiluminescence (Cell Signaling
Technology, Inc.).
Apoptosis assay
Spontaneous apoptosis, as well as apoptosis of cells
in response to selective pharmacological inhibitors, gefitinib
(selective EGFR TKI) and LY294002 (selective PI3K inhibitor), were
examined using Annexin V-PE-Cy5/PI staining (Biosea Biotechnology
Company, China) on a FC500 flow cytometer (Beckman Coulter, Inc.,
CA, USA).
Statistical analysis
Data are presented as the mean ± standard deviation.
One way analysis of variance was used to assess the observed
differences of samples more than two groups. For two groups of
samples, the Student's t-test was used to assess statistical
significance. All statistical tests were two-sided and P<0.05
was considered to indicate a statistically significant
difference.
Results
Generation of cell line with acquired
resistance to gefitinib
The PC9/G gefitinib-resistant cell line was
established by exposing PC9 cells to MNNG and gefitinib. The PC9
and PC9/G cell lines demonstrated significant differences in
gefitinib sensitivity, as indicated by the evaluation of growth
inhibition (Fig. 1A) and apoptosis
in vitro. The IC50 of PC9 cells to gefitinib was
0.06±0.008 µmol/l, while the IC50 of PC9/G cells was
7.29±0.39 µmol/l (Table I). In
addition, the resistance index (IC50 of resistant
cells/IC50 of parent cells) of PC9/G cells was ~138- to
256-fold greater than that of PC9 cells (data not shown). Following
treatment of PC9 and PC9/G cells with gefitinib at a concentration
of 0.1 µmol/l for 72 h, the apoptotic rate of PC9 cells was
26.2±4.55% and that of PC9/G cells was 6.7±0.36% (P<0.05;
Table I).
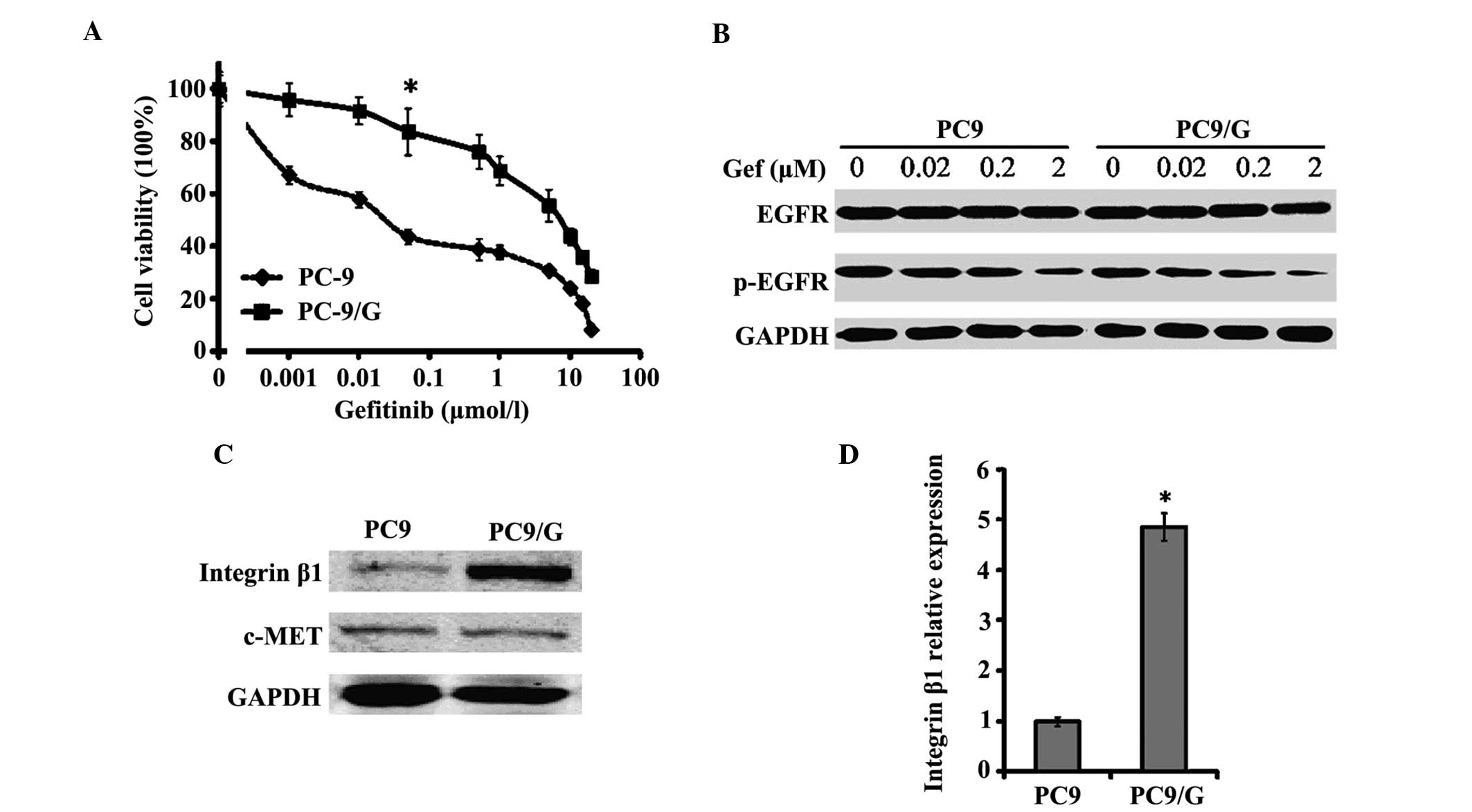 | Figure 1.Characterization of the PC9/G
gefitinib-resistant cell line. (A) Growth-inhibitory effect of Gef
on PC9 and PC9/G cells determined by MTT assay. Cells were seeded
on 96-well plates in quadruplicate, and cultured in the indicated
concentrations of Gef. Following 72 h of incubation, the cells were
subjected to MTT assay. *P<0.05 vs. PC9 cells. (B) Western blot
analysis of EGFR and p-EGFR in PC9 and PC9/G cells. Cells were
placed in medium containing 0, 0.02, 0.2 and 2 µM of gefitinib for
6 h. Samples were separated by SDS-PAGE with a 20 µg sample of cell
lysate loaded into each well. No marked difference in p-EGFR
inhibition was identified between PC9 and PC9/G cells. (C) Western
blot analysis of integrin β1 and c-MET expression in PC9 and PC9/G
cells. A 20 µg sample of cell lysate was separated by SDS-PAGE,
transferred to membranes and incubated with the indicated
antibodies as the first antibody and then with horseradish
peroxidase-conjugated secondary antibody. (D) Polymerase chain
reaction analysis of integrin β1 messenger RNA level in PC9 and
PC9/G cells. *P<0.05 vs. PC9 cells. Gef, gefitinib; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; EGFR,
epidermal growth factor receptor; p, phosphorylated; SDS-PAGE,
sodium dodecyl sulfate polyacrylamide gel electrophoresis. |
 | Table I.Effect of gefitinib with or without
LY294002 on cell proliferation, apoptosis and cell cycle
progression of PC9/G cells. |
Table I.
Effect of gefitinib with or without
LY294002 on cell proliferation, apoptosis and cell cycle
progression of PC9/G cells.
| A, Gefitinib
alone |
|
|
|
|
|---|
|
|
|
|
|
|---|
|
|
|
| Cell cycle phase,
% |
|---|
|
|
|
|
|
|---|
| Cell | IC50
values, µM | Apoptotic rate,
% | G0/G1 | S |
|---|
| PC9 |
0.06±0.008 | 26.2±4.55 |
72.6±5.39 |
4.8±1.6 |
| PC9/G | 7.29±0.39 |
6.7±0.36 | 62.2±3.3 | 15.8±2.8 |
|
| B,
Gefitinib+LY294002 |
|
|
|
|
|
|
|
|
| Cell cycle phase,
% |
|
|
|
|
|
| Cell | IC50
values, µM | Apoptotic rate,
% | G0/G1 | S |
|
| PC9 |
0.05±0.009 | 24.8±2.38 | 75.1±4.3 | 4.5±1.5 |
| PC9/G |
0.092±0.005a |
21.0±0.81a |
76.7±4.6a |
8.9±3.8a |
EGFR gene mutations and levels of
phospho-EGFR are similar in the PC9 and PC9/G cell lines
DNA sequence analysis revealed that the gene status
of EGFR in PC9 and PC9/G cells was identical; the two cell lines
demonstrated depletion of exon 19 and no exon 20 mutations (T790M).
Whether there were any differences in gefitinib-induced inhibition
of EGFR TK was investigated by examining the levels of
phospho-EGFR. It was demonstrated that gefitinib was able to
efficiently induce a similar decrease in phospho-EGFR levels in the
PC9 and PC9/G cell lines (Fig. 1B).
The gefitinib-resistant property of the PC9/G cell line is
therefore not attributable to EGFR mutations or lack of efficacy of
gefitinib on EGFR TK.
Gefitinib inhibits the cell cycle of
PC9 but not PC9/G cells
Cell cycle analysis of the two cell lines was
performed to exclude the possibility of differing cell
proliferation rates between the two cell lines, which may result in
the observed differences in gefitinib sensitivity. When cultured in
10% fetal calf serum-DMEM, the doubling time of PC9/G cells was
20.5 h, which was similar to that of PC9 cells (19.7 h, P>0.05).
There were also no significant differences in the number of cells
at G0/G1 phase and S phase between the PC9/G and PC9 cell lines in
rest state. However, following administration of 0.03 µmol/l
gefitinib, PC9 cells were significantly inhibited at G1 phase
(P<0.05), whereas no notable inhibition of the PC9/G cell cycle
was observed (Table II).
| Table II.Effect of gefitinib on cell cycle
distribution of PC9 and PC9/G cells. |
Table II.
Effect of gefitinib on cell cycle
distribution of PC9 and PC9/G cells.
| A, Cell cycle
analysis prior to gefitinib treatment |
|
|---|
|
|---|
|
| Cell cycle phase,
% |
|---|
|
|
|
|---|
| Cell type | G0/G1 | S | G2/M |
|---|
| PC9 |
49.2±4.1a | 28.3±1.5 | 22.6±2.0 |
| PC9/G | 53.8±4.5 | 23.0±1.2 | 23.2±1.9 |
|
| B, Cell cycle
analysis following gefitinib treatment |
|
|
|
| Cell cycle phase,
% |
|
|
|
| Cell type | G0/G1 | S | G2/M |
|
| PC9 |
87.6±4.3a,b | 2.3±0.6 | 7.5±0.8 |
| PC9/G |
60.9±3.5b | 17.9±1.3 | 21.2±2.1 |
Integrin β1 is highly expressed in
PC9/G cells and its knockdown restores gefitinib sensitivity
Western blot and PCR analyses revealed that there
were significantly higher expression levels of integrin β1 in PC9/G
cells, compared with those of PC9 cells (Fig. 1C and D). In order to determine whether
this abnormally high expression of integrin was associated with the
gefitinib resistance of PC9/G cells, a gene knockdown assay was
conducted by shRNA interference. The efficiency of shRNA
interference was confirmed by western blotting (Fig. 2A). It was demonstrated that knockdown
of integrin in PC9/G cells markedly increased the their sensitivity
to gefitinib treatment, compared with those transfected with
si-scrambled, exhibiting IC50 values of 6.85±0.25 µmol/l
and 0.09±0.006 µmol/l, respectively (P<0.05; Fig. 2B). Furthermore, integrin knockdown did
not affect the spontaneous apoptotic rate of PC9/G cells. Following
gefitinib treatment, the apoptotic rate of PC9/G-si-ITGB1 cells
significantly increased and a greater number of cells were arrested
at G0/G1 phase than in PC9/G or PC9/G-si-scrambled cells (Table III). The observation that integrin
β1 shRNA restored the sensitivity of PC9/G cells to gefitinib
suggested that integrin β1 may mediate the acquired gefitinib
resistance of PC9/G cells.
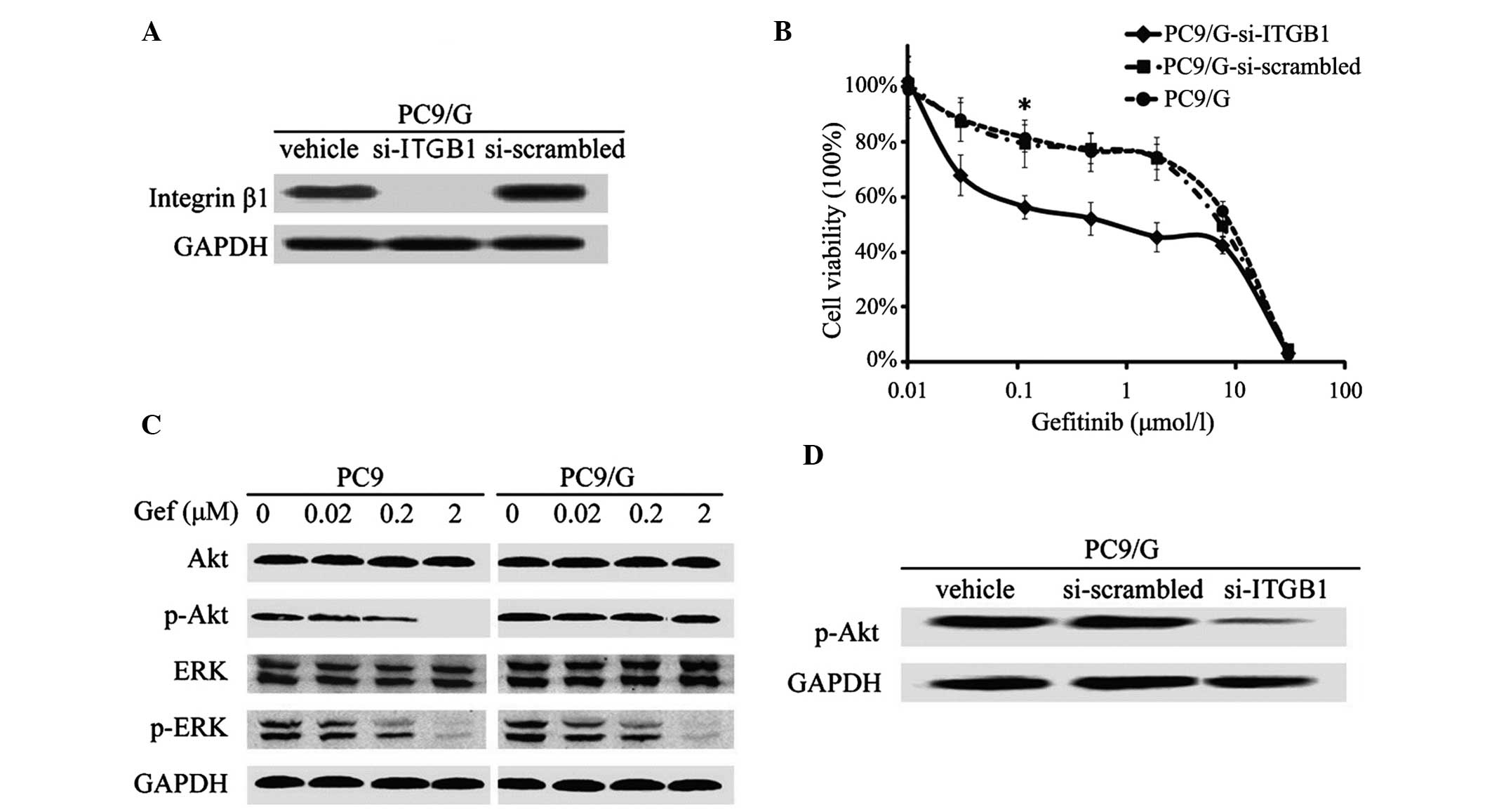 | Figure 2.Characterization of integrin
β1-knockdown and analysis of the downstream pathway of EGFR
signaling in PC9/G cells. (A) Western blot analysis of integrin
expression in PC9/G cells transfected with three types of vector.
Following transient RNA interference, integrin β1 expression was
almost inhibited in PC9/G cells. (B) Growth-inhibitory effect of
Gef on integrin β1-knockdown PC9/G cells determined by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
Following transfection with vectors, PC9/G cells were exposed to
the indicated concentrations of Gef for 72 h. *P<0.05 vs. PC9/G
and PC9/G-si scrambled cells. (C) Western blot analysis of PI3K/Akt
and mitogen-activated protein kinase pathway of EGFR signaling in
PC9 and PC9/G cells. Cells were placed in medium containing 0,
0.02, 0.2 and 2 µM Gef for 6 h and harvested in buffer. Samples (20
µg) were separated by sodium dodecyl sulfate polyacrylamide gel
electrophoresis, transferred to a membrane and blotted with
anti-p-Akt and anti-p-Erk antibodies. (D) Western blot analysis of
activation of PI3K/Akt in integrin-knockdown PC9/G cells. Following
transfection with vectors, PC9/G cells were harvested in buffer.
Total cellular lysates (20 µg) were separated on gel, transferred
to membranes and blotted. Knockdown of integrin β1 induced a
significant decrease in p-Akt expression. EGFR, epidermal growth
factor receptor; Gef, gefinitib; PI3K, phosphoinositide 3-kinase;
ITGB1, p, phosphorylated; integrin β1; si, short interfering; Akt,
protein kinase B; ERK/Erk, extracellular signal-regulated
kinase. |
| Table III.Effect of gefitinib with or without
LY294002 on cell proliferation, apoptosis and cell cycle
progression of PC9/G-si-scrambled/ITGB1 cells. |
Table III.
Effect of gefitinib with or without
LY294002 on cell proliferation, apoptosis and cell cycle
progression of PC9/G-si-scrambled/ITGB1 cells.
| A, Gefitinib
alone |
|---|
|
|---|
|
|
|
| Cell cycle phase,
% |
|---|
|
|
|
|
|
|---|
| Cell | IC50
values, µM | Apoptotic rate,
% | G0/G1 | S |
|---|
|
PC9/G-si-scrambled | 6.85±0.25 |
6.6±0.51 | 58.8±4.6 | 14.9±1.9 |
| PC9/G-si-ITGB1 |
0.09±0.006b |
24.0±3.12b |
75.3±3.5b |
5.1±2.7b |
|
| B,
Gefitinib+LY294002 |
|
|
|
|
|
|
|
|
| Cell cycle phase,
% |
|
|
|
|
|
| Cell | IC50
values, µM | Apoptotic rate,
% | G0/G1 | S |
|
|
PC9/G-si-scrambled |
0.098±0.009a |
22.4±1.10a |
75.4±3.4a |
9.2±2.2a |
| PC9/G-si-ITGB1 | 0.087±0.004 | 26.0±0.91 | 78.8±4.8 | 4.8±1.2 |
Continuous activation of PI3K/Akt in
the gefitinib-resistant PC9/G cell line
The protein expression of c-MET, the amplification
of which is associated with gefitinib resistance, was evaluated. No
significant differences in c-MET protein expression were found
between PC9 and PC9/G cells (Fig.
1C). The potential intracellular signaling pathways mediating
gefitinib resistance in PC9/G cells were therefore further
analyzed. A significant increase in the expression of phospho-Akt
was detected in PC9/G cells, compared with that of PC9 cells, while
the levels of phospho-Erk were similar between the two cell lines
(Fig. 2C). Furthermore, gefitinib
treatment was able to dose-dependently induce an inhibition of Akt
phosphorylation in PC9 cells, but not in PC9/G cells (Fig. 2C).
To explore the underlying mechanism of continuous
activation of PI3K/Akt in PC9/G cells following gefitinib
treatment, the expression profiles of certain associated modulators
were analyzed in the two cell lines. DNA microarray analysis
demonstrated that only expression levels of integrin β1 were found
to be significantly different in the PC9/G cells compared with
those of the PC9 cells. It was therefore hypothesized that the
elevated expression of integrin β1 observed in PC9/G cells may
account for the resistance to EGFR inhibition.
Integrin β1-mediated gefitinib
resistance is dependent on the PI3K/Akt pathway
To explore whether enhanced PI3K signaling was
responsible for integrin β1-mediated resistance to gefitinib, the
correlation between continuous activation of PI3K and increased
expression of integrin β1 was confirmed. It was demonstrated that
knockdown of integrin β1 expression significantly reduced Akt
phosphorylation (Fig. 2D), suggesting
that the high expression levels of integrin β1 may be attributable
to the sustained activity of the PI3K/Akt pathway in PC9/G cells.
PC9/G cells were subsequently treated with gefitinib alone or in
combination with the PI3K inhibitor LY294002. It was demonstrated
that the sensitivity of PC9/G to gefitinib, apoptosis levels and
proportion of cells in G0/G1 phase were significantly increased in
the gefitinib+LY294002 group, suggesting that the restoration of
gefinitib sensitivity PC9/G cells may be PI3K/Akt pathway-dependent
(Table I). Knockdown of integrin β1
expression in PC9/G cells exerted a similar restorative effect as
that of LY294002 (Fig. 2B and D).
Thus, inhibition of PI3K mimicked the sensitivity restoration
effect of integrin β1 knockdown in PC9/G cells. Taken together, the
results of the present study indicate that an integrin-mediated
overactivation of the PI3K/Akt pathway may underlie the mechanism
of gefitinib resistance in PC9/G cells.
Discussion
EGFR-targeted treatment in tumor therapy shows
enhanced clinical efficacy and a reduction in adverse effects,
compared with that of traditional chemotherapy (23,24).
Gefitinib and erlotinib, two small molecular anilinoquinazoline
inhibitors of EGFR, have been approved for use in NSCLC therapy
(25–27). However, the majority of patients who
respond to EGFR TKI therapy will eventually relapse, and the median
time to progression is only 6–12 months (5). The underlying mechanism of this acquired
resistance to EGFR TKIs remains to be fully elucidated.
Integrins are heterodimeric cell surface
glycoproteins, comprised of α and β subunits, which bind ECM
proteins and attach to the actin cytoskeleton within the cell
(13). In addition to their
structural function, integrins also have a crucial role in the
mediation of signal transduction events, which require direct
signaling via integrin-mediated activation of intracellular
signaling cascades or modulation of growth factor-induced signaling
(28). Recently, increasing evidence
has indicated that integrin signaling may function in various
aspects of tumor cell biology. Previous studies have indicated that
integrin signaling blocks drug-induced apoptosis in myeloma,
ovarian cancer and SCLC cells (16,18,19). In
the present study, the hypothesis that integrin may be involved in
mediating gefitinib resistance of NSCLC cells was evaluated. The
PC9/G NSCLC cell line model, with acquired resistance to gefitinib,
was initially established. It was subsequently demonstrated that
integrin β1 signaling inhibited gefitinib-induced apoptosis. It was
also found that integrin was abnormally highly expressed in PC9/G
cells, and that knockdown of integrin was able to effectively
restore the sensitivity of PC9/G cells to gefitinib. These results
indicated that integrin signaling also contributed to the
development of resistance to EGFR-targeted agents, in addition to
previously reported chemotherapeutic drugs, such as paclitaxel and
carboplatin (18,29).
Direct signaling via integrin-mediated activation is
mediated by the activation of intracellular signaling pathways
through binding to tyrosine phosphorylated focal adhesion kinase
(FAK) (30). The most essential
downstream pathways activated by integrin-ECM binding are the
PI3K/Akt and mitogen activated protein kinase pathways, which have
critical roles in tumorigenesis and therapeutic resistance
(31). Transformed cells, including
the majority of lung cancer cell lines, always develop the ability
to survive and proliferate in the absence of cell adhesion
(12). Recently, it was reported that
activation of Akt was able to be induced by integrin β1, in a
manner independent of FAK and EGFR activity (20,32). In
the present study, it was demonstrated that there was a continuous
activation of PI3K/Akt in the gefitinib-resistant PC9/G cell line
and that depletion of integrin β1 by RNAi inhibited this
activation. It was also revealed that integrin β1-mediated
resistance to gefitinib was dependent on the PI3K signaling
pathway.
Akt-mediated phosphorylation negatively regulates
numerous pro-apoptotic factors, including Bad, caspase 9 and the
forkhead transcription factors, and positively regulates survival
factor nuclear factor (NF)-κB; therefore, any of these downstream
molecules may be involved in conferring integrin β1-mediated drug
resistance (33). In the present
study, DNA microarry analysis identified increased expression
levels of NF-κB in PC9/G cells (data not shown). It is possible
that the activation of NF-κB confers a survival advantage to PC9/G
cells treated with gefitinib. In addition, previous studies have
shown that Akt may prevent the release of cytochrome c from
mitochondria, thus contributing to tumor cell survival and
inhibiting apoptosis (11,29,34).
Whether this is the case for gefitinib resistance in PC9/G cell
lines remains to be elucidated.
In addition to direct signaling, integrin-mediated
signal transduction may function via joint integrin/TK receptor
signaling (35). Numerous growth
factor receptors have previously been demonstrated to interact with
integrins to transduce stronger, more efficient signals to
downstream molecules (28,35). It is therefore plausible that the TK
activity of EGFR may be activated via an interaction with integrin,
bypassing the inhibitory effects of EGFR antibody. However, this
mechanism of resistance to EGFR antibody is not possible for
gefitinib, a small molecule EGFR TKI.
Following the generalized clinical application of
EGFR-targeted therapy, increasing numbers of NSCLC patients may be
found to have inherent or acquired resistance. To develop novel
therapeutic strategies for these patients, a full understanding of
the molecular events underlying the resistance to EGFR inhibitors
is required. In the present study, integrin β1-mediated activation
of the PI3K/Akt pathway was identified as one mechanism of survival
and acquired therapeutic resistance to EGFR TKIs in NSCLC cells. It
was therefore hypothesized that co-targeting integrin β1 and EGFR
simultaneously may present a potential strategy to overcome NSCLC
resistance mediated by integrin.
Acknowledgements
The present study was supported by the Natural
Science Foundation of China (grant no. 30873023).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Normanno N, Bianco C, De Luca A and
Salomon DS: The role of EGF-related peptides in tumor growth. Front
Biosci. 6:D685–D707. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maemondo M, Inoue A, Kobayashi K, et al:
Gefitinib or chemotherapy for non-small-cell lung cancer with
mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosell R, Carcereny E, Gervais R, et al:
Spanish Lung Cancer Group in collaboration with Groupe Français de
Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica:
Erlotinib versus standard chemotherapy as first-line treatment for
European patients with advanced EGFR mutation-positive
non-small-cell lung cancer (EURTAC): A multicentre, open-label,
randomised phase 3 trial. Lancet Oncol. 13:239–246. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nguyen KS, Kobayashi S and Costa DB:
Acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small-cell lung cancers dependent on the
epidermal growth factor receptor pathway. Clin Lung Cancer.
10:281–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thomson S, Buck E, Petti F, Griffin G,
Brown E, Ramnarine N, Iwata KK, Gibson N and Haley JD: Epithelial
to mesenchymal transition is a determinant of sensitivity of
non-small-cell lung carcinoma cell lines and xenografts to
epidermal growth factor receptor inhibition. Cancer Res.
65:9455–9462. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Suda K, Tomizawa K, Fujii M, Murakami H,
Osada H, Maehara Y, Yatabe Y, Sekido Y and Mitsudomi T: Epithelial
to mesenchymal transition in an epidermal growth factor
receptor-mutant lung cancer cell line with acquired resistance to
erlotinib. J Thorac Oncol. 6:1152–1161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Onitsuka T, Uramoto H, Nose N, Takenoyama
M, Hanagiri T, Sugio K and Yasumoto K: Acquired resistance to
gefitinib: The contribution of mechanisms other than the T790M,
MET, and HGF status. Lung Cancer. 68:198–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shintani Y, Maeda M, Chaika N, Johnson KR
and Wheelock MJ: Collagen I promotes epithelial-to-mesenchymal
transition in lung cancer cells via transforming growth factor-beta
signaling. Am J Respir Cell Mol Biol. 38:95–104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Damiano JS, Cress AE, Hazlehurst LA, Shtil
AA and Dalton WS: Cell adhesion mediated drug resistance (CAM-DR):
Role of integrins and resistance to apoptosis in human myeloma cell
lines. Blood. 93:1658–1667. 1999.PubMed/NCBI
|
|
12
|
Giancotti FG and Ruoslahti E: Integrin
signaling. Science. 285:1028–1032. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hynes RO: Integrins: Bidirectional,
allosteric signaling machines. Cell. 110:673–687. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
White DE, Kurpios NA, Zuo D, Hassell JA,
Blaess S, Mueller U and Muller WJ: Targeted-integrin in a
transgenic mouse model disruption of human breast cancer reveals an
essential role in mammary tumor induction. Cancer Cell. 6:159–170.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zutter MM, Santoro SA, Staatz WD and Tsung
YL: Re-expression of the alpha 2 beta 1 integrin abrogates the
malignant phenotype of breast carcinoma cells. Proc Natl Acad Sci
USA. 92:7411–7415. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Emmons MF, Gebhard AW, Nair RR, Baz R,
McLaughlin ML, Cress AE and Hazlehurst LA: Acquisition of
resistance toward HYD1 correlates with a reduction in cleaved α4
integrin expression and a compromised CAM-DR phenotype. Mol Cancer
Ther. 10:2257–2266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martin S, Janouskova H and Dontenwill M:
Integrins and p53 pathways in glioblastoma resistance to
temozolomide. Front Oncol. 2:1572012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Scalici JM, Harrer C, Allen A, Jazaeri A,
Atkins KA, McLachlan KR and Slack-Davis JK: Inhibition of α4β1
integrin increases ovarian cancer response to carboplatin. Gynecol
Oncol. 132:455–461. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hodkinson PS, Mackinnon AC and Sethi T:
Extracellular matrix regulation of drug resistance in small-cell
lung cancer. Int J Radiat Biol. 83:733–741. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Velling T, Stefansson A and Johansson S:
EGFR and beta1 integrins utilize different signaling pathways to
activate Akt. Exp Cell Res. 314:309–316. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koizumi F, Shimoyama T, Taguchi F, Saijo N
and Nishio K: Establishment of a human non-small cell lung cancer
cell lines resistant to gefitinib. Int J Cancer. 116:36–44. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Laskin JJ and Sandler AB: Epidermal growth
factor receptor: A promising target in solid tumors. Cancer Treat
Rev. 30:1–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fukuoka M, Yano S, Giaccone G, Tamura T,
Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S,
Rischin D, et al: Multi-institutional randomized phase II trial of
gefitinib for previously treated patients with advanced
non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin
Oncol. 21:2237–2246. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thatcher N, Chang A, Parikh P, Rodrigues
Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH,
Pemberton K, Archer V and Carroll K: Gefitinib plus best supportive
care in previously treated patients with refractory advanced
non-small-cell lung cancer: Results from a randomized,
placebo-controlled, multicentre study (Iressa Survival Evaluation
in Lung Cancer). Lancet. 366:1527–1537. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shepherd FA, Rodrigues Pereira J, Ciuleanu
T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S,
Smylie M, Martins R, et al: National Cancer Institute of Canada
Clinical Trials Group: Erlotinib in previously treated
non-small-cell lung cancer. N Engl J Med. 353:123–132. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Parise LV, Lee J and Juliano RL: New
aspects of integrin signaling in cancer. Semin Cancer Biol.
10:407–414. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aoudjit F and Vuori K: Integrin signaling
inhibits paclitaxel-induced apoptosis in breast cancer cells.
Oncogene. 20:4995–5004. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Plopper GE, McNamee HP, Dike LE,
Bojanowski K and Ingber DE: Convergence of integrin and growth
factor receptor signaling pathways within the focal adhesion
complex. Mol Biol Cell. 6:1349–1365. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee HY, Srinivas H, Xia D, Lu Y, Superty
R, LaPushin R, Gomez-Manzano C, Gal AM, Walsh GL, Force T, et al:
Evidence that phosphadidylinositol 3-kinase- and mitogen-activated
protein kinase kinase-4/c-Jun NH2-terminal kinase-dependent
pathways cooperate to maintain lung cancer cell survival. J Biol
Chem. 278:23630–23638. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zeller KS, Idevall-Hagren O, Stefansson A,
Velling T, Jackson SP, Downward J, Tengholm A and Johansson S:
PI3-kinase p110α mediates β1 integrin-induced Akt activation and
membrane protrusion during cell attachment and initial spreading.
Cell Signal. 22:1838–1848. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gilmore AP, Metcalfe AD, Romer LH and
Streuli CH: Integrin-mediated survival signals regulate the
apoptotic function of bax through conformation and subcellular
localization. J Cell Biol. 149:431–446. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sethi T, Rintoul RC, Moore SM, MacKinnon
AC, Salter D, Choo C, Chilvers ER, Dransfield I, Donnelly SC,
Strieter R and Haslett C: Extracellular matrix proteins protect
small cell lung cancer cells against apoptosis: A mechanism for
small cell lung cancer growth and drug resistance in vivo. Nat Med.
5:662–668. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cabodi S, Morello V, Masi A, Cicchi R,
Broggio C, Distefano P, Brunelli E, Silengo L, Pavone F, Arcangeli
A, et al: Convergence of integrins and EGF receptor signaling via
PI3K/Akt/FoxO pathway in early gene Egr-1 expression. J Cell
Physiol. 218:294–303. 2009. View Article : Google Scholar : PubMed/NCBI
|














