Introduction
The daily light/dark cycles of the Earth are
responsible for the physiological and behavioral activity of a
number of organisms. This temporal activity is known as circadian
rhythm, and has a biological molecular basis, namely the circadian
gene (1,2). Previous studies have demonstrated that
the circadian genes regulate several molecular and biochemical
processes as well as having an established role in the mammalian
circadian clock. Research suggests that the role of the circadian
clock may be a fundamental regulator for tumor suppression in
humans (3,4). Several cancer studies indicate that
period 2 (Per2), one of the key circadian genes, plays a
significant role in growth control and tumor development (5). To understand the effects of Per2 on
osteosarcoma cell growth in vitro, the recombinant
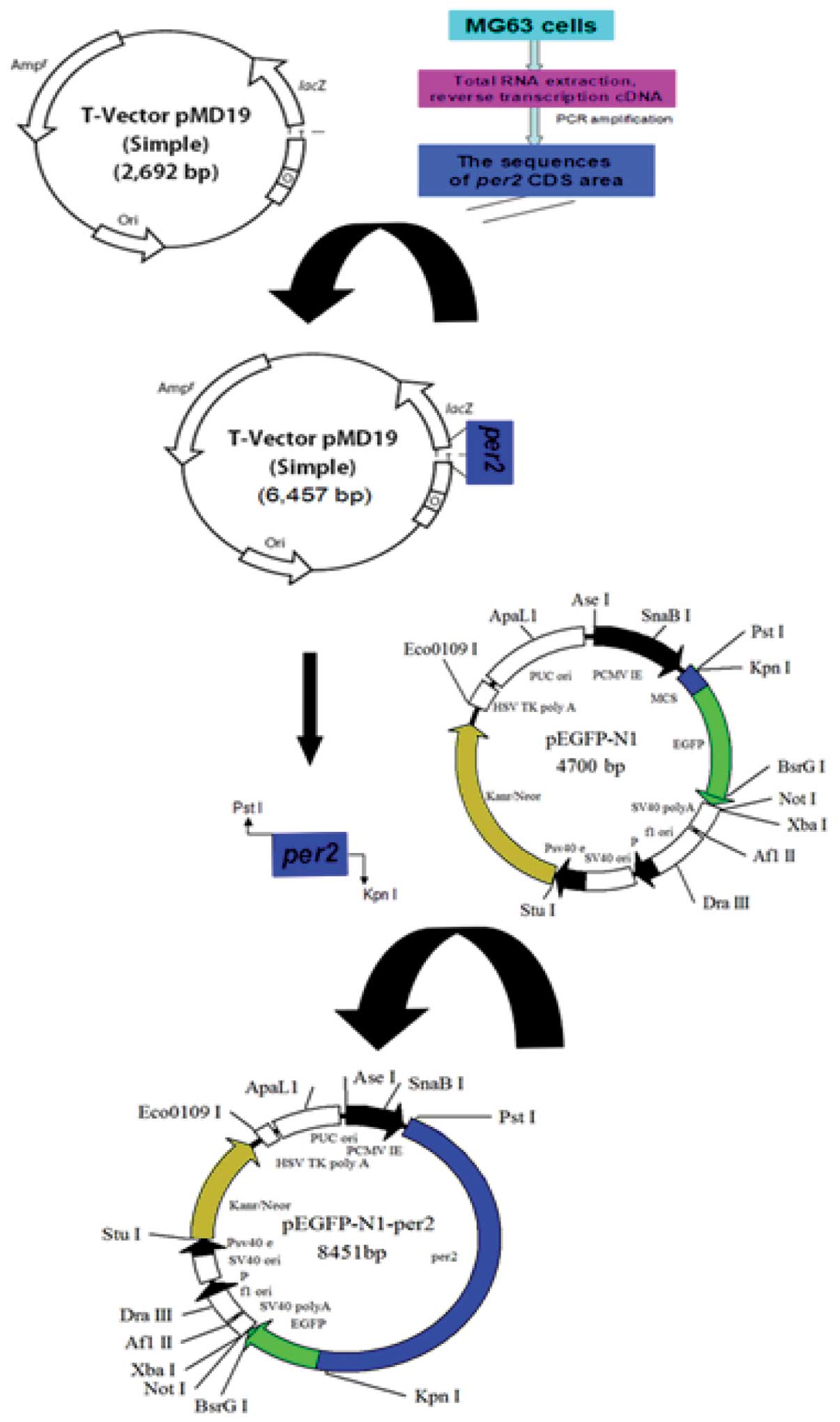
pEGFP-N1-hPer2 plasmid was constructed using the vector pEGFP-N1
carrying the fluorescent protein gene and transfected into MG63
cells using Lipofectamine 2000, then the expression of hPer2 in
MG63 cells was assessed by reverse transcription-polymerase chain
reaction and western blot analysis. This research is expected to
lay the foundations for research into the circadian gene in
osteosarcoma.
Materials and methods
Materials
The MG63 osteosarcoma cell line was purchased from
the Cell Bank of Wuhan University (Wuhan, China); the pMD19-T
vector, Escherichia coli DH5a and DNA marker were purchased
from Transgen Biotechnology (Beijing, China); PstI and
KpnI restriction enzymes were purchased from NEB (Ipswich,
MA, USA); the plasmid pEGFP-N1 was purchased from Clonetech
Biotechnology (Mountain View, CA, USA); the DNA ligation kit,
RevertAid™ First Strand cDNA synthesis kit, dNTPs, RevertAid
reverse transcriptase and HiFi DNA polymerase were purchased from
Fermentas (Beijing, China); TRIzol™ reagent, Lipofectamine 2000 and
radioimmunoprecipitation assay (RIPA) lysis buffer were purchased
from Beyotime Institute of Biotechnology (Shanghai, China); AxyPrep
DNA gel extraction kit was purchased from Takara Biotechnology,
Inc. (Dalian, China); rabbit anti-hPer2 polyclonal antibody was
purchased from ProteinTech Group, Inc. (Chicago, IL, USA; catalog
no., 20359–1-AP); and Western Lighting Plus Chemiluminescence was
purchased from PerkinElmer, Inc. (Waltham, MA, USA).
Methods
Gene amplification, cloning and sequencing
Total RNA was extracted from the osteosarcoma cell
line MG63 using TRIzol reagent according to the procedure supplied
by the manufacturer. Extracted RNA (1 µg) was used for cDNA
synthesis with the RevertAid First Strand cDNA synthesis kit
according to the manufacturer's instructions. The reaction system
was prepared in a total volume of 20 µl containing 12.5 µl RNA
primer mix, 4 µl 5X RT reaction buffer, 2 µl dNTPs, 1 µl RevertAid
reverse transcriptase, 0.5 µl RiboLock RNase inhibitor and
dH2O up to 20 µl. A pair of primers was designed based
on the hPer2 mRNA sequence (Genbank no. NM_022817.2): PstI
tailed forward (5′-AACTGCAGATGAATGGATACGCGGAATTTCC-3′) and
KpnI tailed reverse (5′-CGGCTGCAGCGTCTGCTCTTCGATCCTGT-3′)
primers (restriction sites are underlined) and named as hPer2-F and
hPer2-R, respectively. The length of the amplification segment was
3765 bp. The PCR mixture was blended in a total volume of 20 ml
containing 2 µl cDNA template, 1 µl each primer (10 µmol/l), 10 µl
PCR mix and dH2O up to 20 µl. The PCR program was
started at 94°C for 5 min, followed by 40 cycles at 94°C for 30
sec, 58°C for 30 sec, 72°C for 5 min and completed with a final
extension at 72°C for 5 min. The final PCR products were separated
by electrophoresis using 1% polyacrylamide gels, and the target
fragment was purified and recovered using an agarose gel extraction
kit (Watson Biomedical, Inc., Shanghai, China).
The purified target fragments were ligated into the
plasmid pMD19-T and then transformed into competent E.coli
DH5a cells. Recombinant plasmid was extracted from bacterial
colonies and 1.0 µl plasmid solution was subjected to agarose gel
electrophoresis to confirm the presence of the correct sequence of
hPer2.
Construction of pEGFP-N1-hPer2 expression
plasmid
Double restriction enzyme digestion was applied to
the recovered target gene fragment and eukaryotic expression vector
pEGFP-N1, respectively. The enzyme reaction contained 2 µl target
gene fragment (or vector pEGFP-N1), 2 µl 10X Fast Digest buffer, 1
µl PstI, 1 µl KpnI and dH2O up to 20 µl.
Under the guidance of the T4 DNA ligase system instructions, the
purified target fragment of the hPer2 gene was directionally
ligated into pEGFP-N1 vector in a 10-µl reaction system containing
3 µl target fragment, 1 µl pEGFP-N1, 1 µl T4 DNA ligase, 1 µl 2X
Quick Ligation buffer and dH2O up to 10 µl. The
reactants were well mixed at 16°C for 2 h, then the ligation was
transformed into competent E.coli DH5a cells and inoculated into
Luria-Bertani culture media containing 100 µg/ml ampicillin at a
volume ratio of 1:100. After amplification by shaking the culture
overnight at 37°C, the target plasmids were extracted from the
bacterial liquid according to the instructions for the EndoFree
Maxi Plasmid kit (Tiangen Biotech Co., Ltd., Beijing, China). The
resulting recombinant eukaryotic expression vector was named
pEGFP-N1-hPer2, and the construction procedure is shown in Fig. 1.
The pEGFP-N1-hPer2 was digested using PstI
and KpnI, and then evaluated by agarose gel electrophoresis.
The recombinant plasmid was further sequenced to confirm its
sequence by the Beijing Genomics Institute (BGI; Beijing,
China).
Transfection of pEGFP-N1-hPer2 into MG63
cells
MG63 cell lines originated from human osteosarcoma
were used in this study. The cells were maintained and cultured in
RPMI-1640 media supplemented with 10% fetal bovine serum, 100 U/ml
penicillin and 100 µg/ml streptomycin (Biochrom KG, Berlin,
Germany) at 37°C in a humidified 5% CO2 incubator. The
cells were divided into three groups: pEGFP-hPer2, pEGFP-N1 and
control. Cells were transiently transfected with the DNA construct
using Lipofectamine 2000 reagent. In brief, following the
manufacturer's instructions the transfection complex was prepared
based on the optimized amounts of plasmid and Lipofectamine reagent
and transferred to the 70–80% confluent MG63 cells, then cells were
washed with phosphate-buffered saline (PBS) and collected to
conduct subsequent assays.
Analysis of transfection efficiency
The expression of enhanced green fluorescence
protein (EGFP) was already visualized by fluorescence microscopy
(Olympus IX51; Olympus Corporation, Seoul, South Korea) at 24 h
post-transfection, and the percentage of fluorescence-emitting
cells was determined by flow cytometry (FACSort; BD Biosciences,
Franklin Lakes, NJ, USA).
Quantitative RT-PCR (RT-qPCR) analysis
Total RNA was isolated from MG63 cells using TRIzol
reagent, and the concentration of each RNA sample was determined
using a NanoVue Plus spectrophotometer (GE Healthcare Bio-Sciences
AB, Uppsala, Sweden). All RNA samples were subsequently adjusted to
the same concentration. An SYBR PrimeScript RT-PCR kit (Takara
Biotechnology, Inc.) was then used for RT-PCR according to the
manufacturer's instructions. The relative mRNA expression of hPer2
was analyzed by qPCR using the IQ™5 System (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) with β-actin (Genbank no. NM_001101)
serving as the reference gene. The primer information is as
follows: hPER2-F, TACACCGTGGAGGAGATGGAGA; hPER2-R,
ATATGGATGCAACCTGGTCAGA; β-actin-F, GTCCACCGCAAATGCTTCTA; β-actin-R,
TGCTGTCACCTTCACCGTTC. The PCR reactions were carried out in a
96-well plate in a 25 µl reaction volume. Each reaction mixture
contained 12.5 µl SYBR-Green I PCR Master mix (Takara
Biotechnology, Inc.), 2.5 µl normalized template DNA, 0.5 µl each
primer and 9.5 µl sterile ultrapure water. The relative expression
of hPer2 was calculated using the ‘normalized relative
quantification’ method followed by the 2−∆∆Ct cycle
threshold method (6). PCR reactions
were performed in triplicate for each sample.
Western blot analysis
For western blot analysis, cells at 90% confluency
were washed in PBS before incubation with RIPA lysis buffer
consisting of 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1%
sodium deoxycholate, 0.1% sodium dodecyl sulphate (SDS) on ice for
10 min. The cell lysates were clarified by centrifugation at 9000 ×
g for 10 min, and the supernatants were collected. Protein
concentration was measured by bicinchoninic assay (Aidlab, Beijing,
China). Equal amounts of total protein were separated on 10%
SDS-polyacrylamide gel, and then transferred electrophoretically to
nitrocellulose membranes blocked with TBST buffer (50 mM Tris-HCl,
pH 7.5, 0.15 M NaCl, 0.1% Tween-20) containing 5% fat-free dry milk
for 2 h and incubated for 3 h with rabbit polyclonal anti-human
hPer2 antibody (dilution, 1:500) in TBST. Following incubation with
a horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (dilution, 1:2000; catalog no., SA00001-2; ProteinTech
Group, Inc.), immunoreactive proteins were visualized with an
enhanced chemiluminescence detection system. The western blot
experiments were repeated three times. The relative expression of
the target protein was calculated as the gray value ratio of target
protein content to β-actin content (target protein/β-actin) using
Quantity One version 4.62 image analysis software (Bio-Rad
Laboratories, Inc.).
Statistical analysis
Statistical analyses were carried out with the SPSS
version 17.0 (SPSS, Inc., Chicago, IL, USA) statistical software
package for Windows. All analyses in this study were performed
using analysis of variance. All P-values were based on two-tailed
tests and P<0.05 was considered to indicate a statistically
significant difference.
Results
Agarose gel electrophoresis of RT-PCR
product
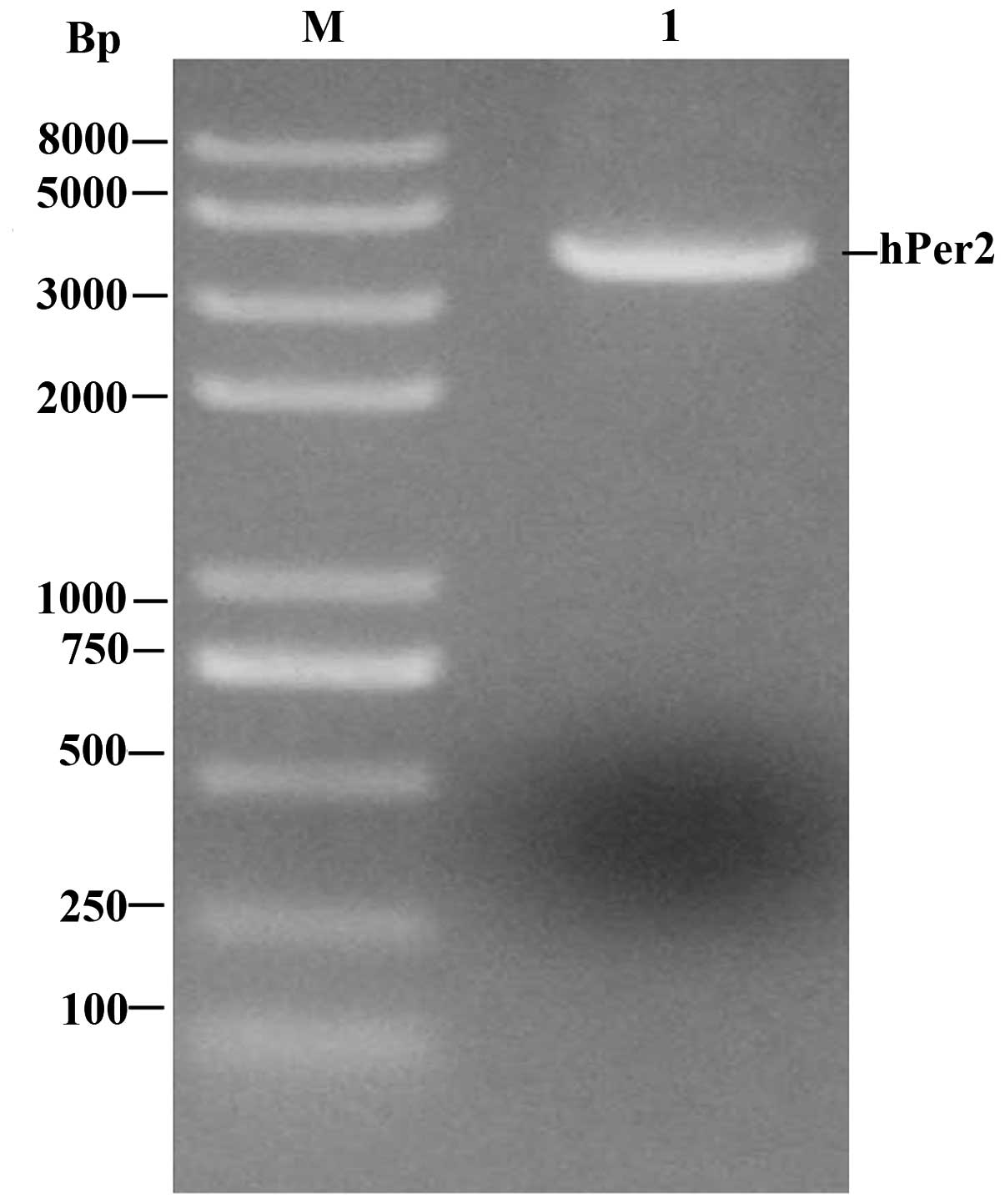
The results of RT-PCR demonstrated that there was a
visible DNA band just below 4 kb which had the same size as the
expectant target gene (the length of hPer2 was 3765 bp; Fig. 2).
Identification of recombinant
expression vector
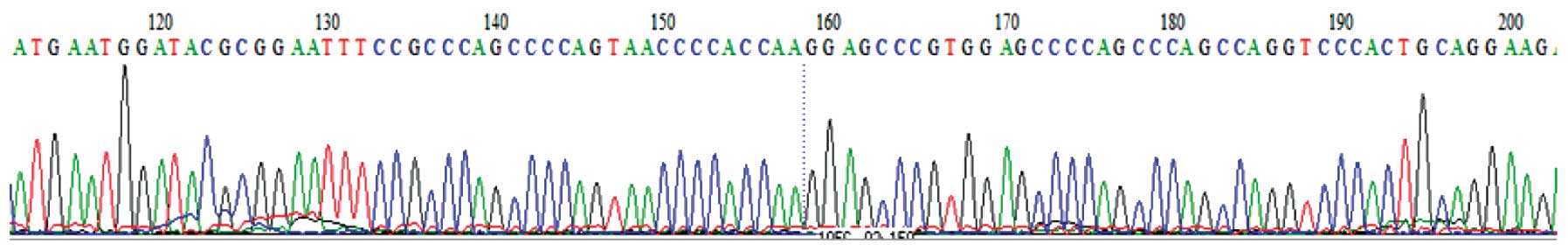
The DNA sequencing results revealed that the
sequence of the 3765 bp inserted segment was identical to the cDNA
sequence of the hPer2 gene. The results of nucleotide-nucleotide
BLAST in NCBI also demonstrated that the sequence alignment was
completely correct (Fig. 3).
Following double enzyme digestion, a 3765-bp insertion segment and
a 4.7-kb vector fragment were observed in the pEGFP-N1-hPer2 group
following electrophoresis, while only a 4.7-kb vector fragment was
observed in the pEGFP-N1 group. The results confirmed that the
construction of the pEGFP-N1-hPer2 eukaryotic expression plasmid
was successful (Fig. 4).
Transfection efficiency analysis of
hPer2 gene in MG63 cells in vitro
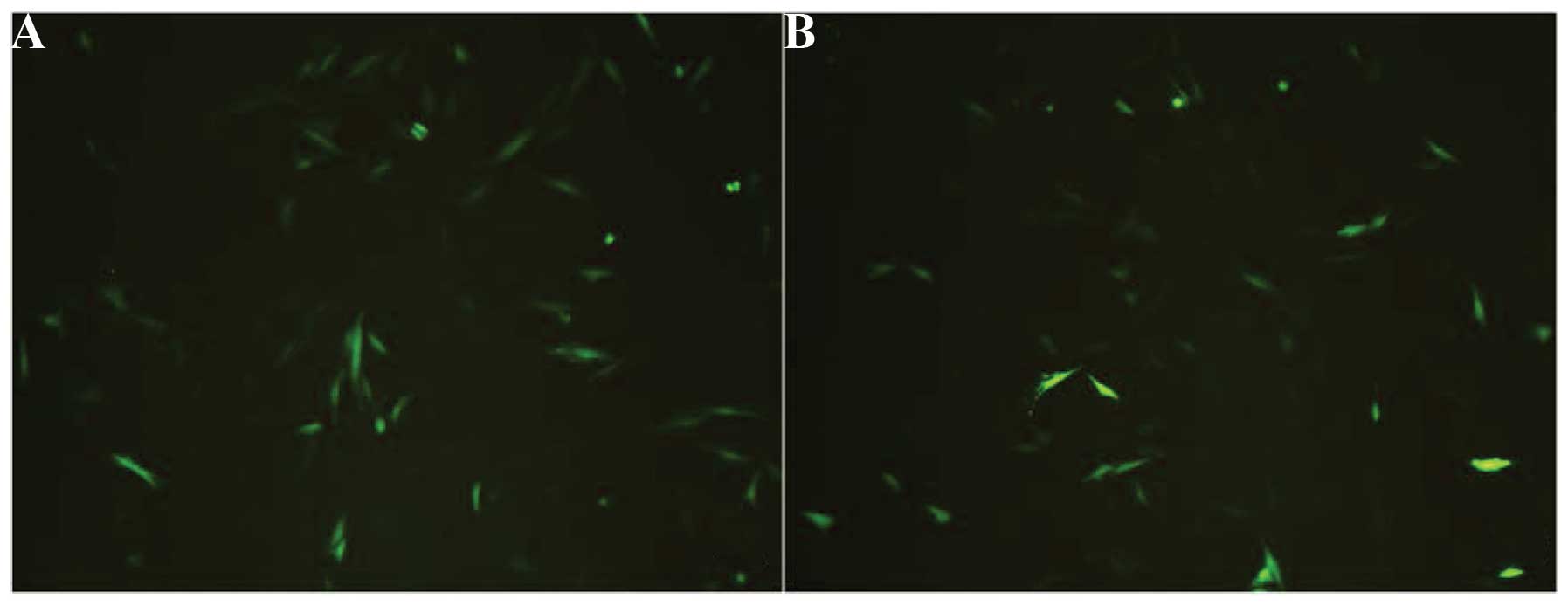
The expression of the EGFP reporter gene was clearly
observed using fluorescence microscopy (Olympus IX51) 48 h after
transfection (Fig. 5) in the
pEGFP-N1-hPer2 and the pEGFP-N1 group but not in the control group.
The results revealed that in the pEGFP-N1-hPer2 and the pEGFP-N1
group, large numbers of MG63 expressed GFP. EGFP was expressed in
70% of cells in the pEGFP-N1-hPer2 group and 75% of cells in the
pEGFP-N1 group, suggesting that pEGFP-N1-hPer2 and pEGFP-N1 may be
effectively transfected into MG63 cells, resulting in a high level
of EGFP expression. It was expected that the pEGFP-N1 group would
express a more intense fluorescence signal than the pEGFP-N1-hPer2
group, since the empty pEGFP-N1 vector was smaller and had a higher
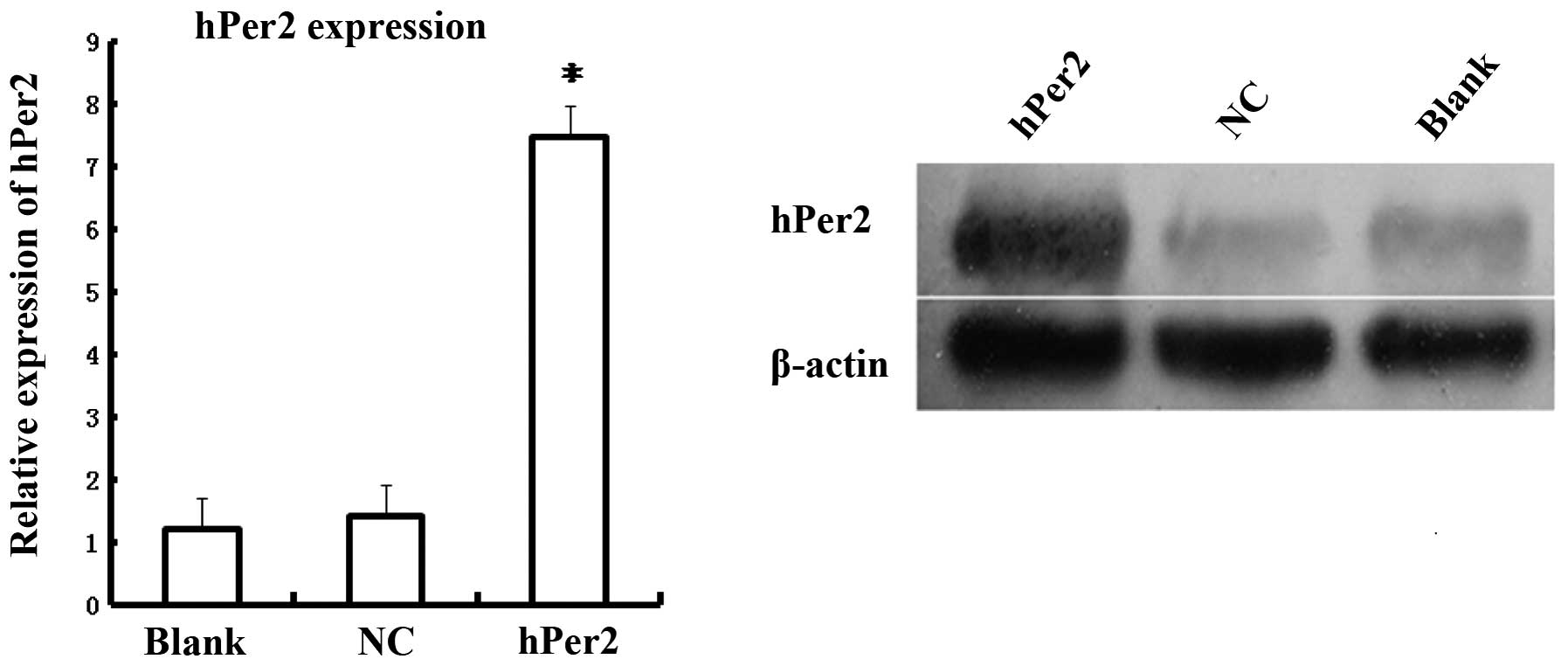
transfection efficiency. In addition, as shown in Fig. 6, hPer2 mRNA expression was
significantly higher in the pEGFP-N1-hPer2 group compared with the
pEGFP-N1 or control groups (P<0.05), and no statistical
difference existed between pEGFP-N1 and the control group
(P>0.05), suggesting that hPer2 was successfully transfected
into MG63 cells and efficiently expressed. Western blot analysis
revealed that hPer2 protein exhibited a significant upregulation in
the pEGFP-N1-hPer2 group compared with the pEGFP-N1 and control
group (P<0.05), and no statistical difference existed between
pEGFP-N1 and the control group (P>0.05). These data also
demonstrated that human osteosarcoma MG63 cell lines were
successfully generated, in which hPer2 was overexpressed.
Discussion
Osteosarcoma is the most common primary bone tumor
and mainly affects children and adolescents (7,8). The
etiology of osteosarcoma is largely unknown due to the difficulties
in understanding the molecular mechanism of tumor development in
the complex structure and numerous genomic rearrangements of bone
cancer cells (9). Complete radical
surgery remains a preferable choice in osteosarcoma treatment, with
adjuvant chemotherapy administered prior to surgery (10). If surgical excision is not possible,
the addition of radiation therapy may be beneficial to control the
local tumor. Still, a number of patients with osteosarcoma risk
having local relapse following chemotherapy (11). For this reason, it is crucial to
explore novel and alternative strategies for osteosarcoma
treatment. Understanding the fundamental molecular mechanisms in
the pathogenesis of osteosarcoma may help to develop novel
strategies that have a specific molecular target for the treatment
of patients with osteosarcoma (12).
Life on earth has evolved in the presence of a
rhythmically changing environment. Most eukaryotes, and certain
prokaryotes, have developed a molecular time-keeping mechanism that
synchronizes itself with the external environment to ensure optimal
timing of cellular functions, metabolism and physiology (13–15). This
mechanism is known as the circadian oscillator or clock. Circadian
clocks have been identified in the majority of tissues and cells of
mammals (16,17). The molecular basis of the circadian
clock is the oscillatory transcription and translation of ‘clock
genes’. Studies suggest that tumorigenesis is associated with
altered circadian function, whether causal or symptomatic, and that
a dysfunctional circadian clock promotes carcinogenesis (18). However, the effects of clock genes on
the biological behavior of osteosarcoma cells are rarely reported.
In the current study, we selected hPer2, one of the key circadian
genes, and successfully constructed the recombinant pEGFP-N1-hPer2
plasmid, which was transfected into the MG63 osteosarcoma cell
line. This study is expected to lay the foundation for research
into the circadian gene in osteosarcoma. A gene transformation
technique is applied to modify tumor cells with certain functional
genes to increase inhibition effects on the growth of tumors. The
development of this technique has been extremely rapid in recent
years and has become a new method in the precaution and treatment
of tumors (19).
GFP easily forms a fusion protein with other target
genes and has no influence on the space conformation and function
of the target gene product. It has become the optimal reporter gene
for the detection of the target gene transfection efficiency and
expression modality (20). EGFP is
optimized mutant GFP possessing a much higher sensitivity and 35
times stronger fluorescence than the wild type, having no species
specificity and a notable influence on the growth and function of
cells (21).
Taken together, in this study, in order to detect
the transfection efficiency of the hPer2 gene, we constructed a
recombinant plasmid using EGFP which exhibits the changes of its
expression position and quantity within MG63 cells. We investigated
the function of the target gene in the occurrence and development
of tumors, as well as the molecular mechanisms involved. Our
preliminary studies provide the ground work for further research on
the roles of the circadian gene hPer2 in MG63 osteosarcoma cells,
and pave the way for future studies of the intracellular downstream
signaling mechanisms responsible for hPer2′s ability to affect
osteosarcoma cells.
Acknowledgements
The authors thank the Central Laboratory of the
First Affiliated Hospital of Wuhan University, as well as Jishuang
Zhu for his assistance in primer design and Dr Ling Yu for his
careful review of the manuscript.
References
|
1
|
Partch CL, Green CB and Takahashi JS:
Molecular architecture of the mammalian circadian clock. Trends
Cell Biol. 24:90–99. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kelleher FC, Rao A and Maguire A:
Circadian molecular clocks and cancer. Cancer Lett. 342:9–18. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greene MW: Circadian rhythms and tumor
growth. Cancer Lett. 318:115–123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beckett M and Roden LC: Mechanisms by
which circadian rhythm disruption may lead to cancer. S Afr J Sci.
105:415–420. 2009.
|
|
5
|
Ishida N: Circadian clock, cancer and
lipid metabolism. Neurosci Res. 57:483–490. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Admassi D: Osteosarcoma of medial cuniform
bone. Ethiop Med J. 47:305–308. 2009.PubMed/NCBI
|
|
9
|
Martin JW, Squire JA and Zielenska M: The
genetics of osteosarcoma. Sarcoma. 13:6272542012.
|
|
10
|
Yamamoto N and Tsuchiya H: Chemotherapy
for osteosarcoma - where does it come from? What is it? Where is it
going? Expert Opin Pharmacother. 14:2183–2193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luetke A, Meyers PA, Lewis I and Juergens
H: Osteosarcoma treatment - where do we stand? A state of the art
review. Cancer Treat Rev. 40:523–532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang J and Zhang W: New molecular insights
into osteosarcoma targeted therapy. Curr Opin Oncol. 25:398–406.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McWatters HG, Roden LC and Staiger D:
Picking out parallels: Plant circadian clocks in context. Philos
Trans R Soc Lond B Biol Sci. 356:1735–1743. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levine JD: Sharing time on the fly. Curr
Opin Cell Biol. 16:210–216. 2006. View Article : Google Scholar
|
|
15
|
Dong G and Golden SS: How a cyanobacterium
tells time. Curr Opin Microbiol. 11:541–546. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoo SH, Yamazaki S, Lowrey PL, Shimomura
K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, et al: PERIOD
2: LUCIFERASE real-time reporting of circadian dynamics reveals
persistent circadian oscillations in mouse peripheral tissues. Proc
Natl Acad Sci USA. 101:5339–5346. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peirson SN, Butler JN, Duffield GE, Takher
S, Sharma P and Foster RG: Comparison of clock gene expression in
SCN, retina, heart and liver of mice. Biochem Biophys Res Comm.
351:800–807. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Noda M, Takahashi C, Matsuzaki T and
Kitayama H: What we learn from transformation suppressor genes:
lessons from RECK. Future Oncol. 6:1105–1116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hoffman RM: Imaging metastatic cell
trafficking at the cellular level in vivo with fluorescent
proteins. Methods Mol Biol. 1070:171–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sakharova NY, Mezhevikina LM, Smirnov AA
and Vikhlyantseva EF: Analysis of the effects of blue light on
morphofunctional status of in vitro cultured blastocysts from mice
carrying gene of enhanced green fluorescent protein (EGFP). Bull
Exp Biol Med. 157:162–166. 2014. View Article : Google Scholar : PubMed/NCBI
|