Introduction
Radiotherapy (RT) is commonly used to treat
multi-cancers, including colorectal, lung, prostate, and breast
cancer (1). Radiation may be used
prior to or following surgical resection of cancerous tumors to
attenuate the risk of recurrence and it is often combined with
chemotherapy. While a proportion of tumors do respond well
initially, a large proportion of patients experience resistance to
RT (2). Therefore, it is important to
elucidate the molecular basis that contributes to RT resistance in
order to screen patients prior to them receiving therapy. In
addition, the development of adjuvant treatments to enhance the
efficacy of radiation are imperative.
Radiation treatment generates free radicals and
reactive oxygen species (ROS) that attack the covalent bonds of
DNA, leading to breaks in double stranded DNA (DSBs) (3). In response to DNA damage, the kinase
Ataxia Telangiectasia Mutated (ATM) is activated through
auto-phosphorylation to stimulate DNA repair pathways (4). ATM also regulates the pro-survival and
radio-resistance pathway of Akt-mammalian target of rapamycin
(mTOR), through which the cell survival pathway is activated
(5).
The energy sensor adenosine monophosphate-activated
kinase (AMPK) is a heterotrimeric enzyme composed of one catalytic
subunit and two regulatory subunits with critical roles in
regulating growth and reprogramming metabolism (6). It is a downstream effector of liver
kinase-B 1 (LKB1), a tumor suppressor gene that is mutated in
Peutz-Jeghers syndrome (7). AMPK is
also a highly conserved sensor of intracellular adenosine
nucleotide levels; under energy stress, AMPK is activated through
the phosphorylation on Thr172 by LKB1 (8) to restrict energy consuming anabolic
processes such as protein synthesis, cell cycle and proliferation
instead of the stimulation of substrate uptake and energy
generation through processes such as glucose and amino acid uptake.
Moreover, AMPK induces cell cycle arrest in response to metabolic
stress through induction of p53 (9).
It has been reported that AMPK inhibits mTORC1 through
phosphorylation of tuberous sclerosis 2 (TSC2) on Ser1387 and
phosphorylation of Raptor, an essential component of mTORC1
(10). A recent study demonstrated
that AMPK may be activated by irradiation in an LKB1-independent
manner through the ATM-AMPK-p53/p21 cip1 signaling pathway that
facilitates the irradiation-induced cell cycle arrest at G2-M phase
(11), indicating that targeting the
AMPK pathway may be a novel strategy for radio-sensitization in
human cancer. In the present study, AMPK expression levels were
compared in radiation resistant and radiation sensitive colorectal
cancer patients. By establishing the radiation resistant cell line
from colon cancer cells, the AMPK expression levels were determined
in radioresistant cells and the role of AMPK inhibitor in reversing
the radioresistance was also investigated.
Materials and methods
Patients and sample
A total of 5 fresh tissue samples, from radiation
sensitive and resistant colon cancers, were procured from surgical
resection specimens collected by the Division of General Surgery,
China Japan Union Hospital of Jilin University (Changchun, China)
from March 2012 to May 2013. Primary tumor regions and
corresponding histologically normal tissues from the same patients
were separated by experienced pathologists, and immediately stored
in liquid nitrogen (−193°C) until use. The use of patient samples
was approved by the institutional ethics committee of Radiation
Medicine Institute.
Cells culture and ionizing
radiation
The human colon cancer cell line DLD-1 was purchased
from the American Type Culture Collection (Manassas, VA, USA).
Cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) with added 10% fetal bovine serum (Thermo
Fisher Scientific, Inc.), 2 mM glutamine (Thermo Fisher Scientific,
Inc.), 50 IU/ml penicillin (Thermo Fisher Scientific, Inc.) and 50
µg/ml streptomycin (Thermo Fisher Scientific, Inc.), and were
maintained in humidified 37°C, 5% CO2 incubators. Prior
to collection, cultures were tested for mycoplasma infection using
Myco Alert (Lonza, Walkersville, MD, USA) according to the
manufacturer's protocol. Cells were exposed to different doses of
irradiation (IR) using a 60 Co clinical radiation unit (Co-60
T780C; Best Theratronics, Ltd., Kanata, Canada). Cells were
pre-incubated with drugs (Metformin, 0.5, 1 and 2 µM; Compound C,
1, 2, 4 and 8 µM) for 1 h before IR, followed by incubation for 1 h
at 37 °C 5% CO2 incubators, then the cells were
subjected to downstream analysis. Cells were repeatedly exposed to
gradually increasing doses of IR from 1 to 20 Gy for 4 months, and
the surviving cells were collected. Radioresistant DLD-1 cell
clones were pooled for the following experiments.
Antibodies and reagents
Antibodies used for this study were: mouse
monoclonal AMPKα1 Antibody (H-4) (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA; sc-398861); rabbit polyclonal p-AMPKα1/2 Antibody
(Thr 172) (Santa Cruz Biotechnology, Inc.; sc-33524); mouse
monoclonal GAPDH (Santa Cruz Biotechnology, Inc.; sc-365062).
Metformin and Compound C were purchased from Sigma-Aldrich
(Shanghai, China). The secondary antibodies used were as follows:
Goat anti-rabbit immunoglobulin (Ig)G, horseradish peroxidase
(HRP)-linked antibody (Cell Signaling Technology, Inc., Danvers,
MA, USA; 7074) and horse anti-mouse IgG HRP-linked antibody (Cell
Signaling Technology, Inc.; 7076).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was isolated from cultured cells using the
RNeasy mini-kit (Qiagen GmbH, Hilden, Germany) (with an on-column
DNAse digestion step according to the manufacturer's instructions).
Briefly, lysates of cells were passed through a Qiashredder (Qiagen
GmbH) and the eluted lysates were mixed 1:1 with 70% ethanol. The
lysates were applied to a mini-column and after washing and DNAse I
digestion, the RNA was eluted in 30–50 µl of RNAse-free water. The
quantity and quality of total RNA samples was checked by
agarose-gel-electrophoresis and the Bioanalyzer RNA 6000 Nano assay
(Agilent Technologies GmbH, Waldbronn, Germany). The sequences of
the primers used for RT-qPCR were as follows: AMPKα1, F
5′-CTCAGTTCCTGGAGAAAGATGG-3′ and R 5′-CTGCCGGTTGAGTATCTTCAC-3′; and
18S rRNA, F 5′-TGCTGTCCCTGTATGCCTCT-3′ and R
5′-TGTAGCCACGCTCGGTCA-3′.
Plasmid DNA and siRNA
transfections
Transfection was performed using the Lipofectamine
2000 Transfection reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Briefly, 12 h prior
to transfection, cells were switched to medium without antibiotics.
The transfections were performed when cells reached 80% confluence,
using a 1:3 ratio of DNA (µg) to Lipofectamine (µl); medium was
switched to regular medium 12 h following transfection.
Overexpression vectors containing wild type AMPKα1 were obtained
from OriGene Technologies, Inc. (Rockville, MD, USA). siRNA for
scramble control and siAMPKα1 were also purchased from Origene
Technologies, Inc. The cells were collected or whole-cell lysates
were prepared 48 h after transfection, for further analysis.
Cell viability assay
A total of 5×105 cells/well were seeded
in 6-well plates and incubated overnight. The medium was replaced,
then the cells were treated with either Metformin or Compound C for
1 h at the indicated concentrations followed by exposure to
radiation (Metformin treatment: 0, 1, 2, 4, 8 or 16 Gy; Compound C
treatment: 0, 0.5, 1, 2, 4 or 8 Gy). The cell viability was
determined by trypan blue exclusion test with trypan blue (0.4%)
purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Western blot analysis
Whole cell extracts were prepared from cultured
cells by homogenizing cells in a lysis buffer (10 mM Tris-HCl (pH
7.5), 150 mM NaCl, 1% NP40) containing Halt™ Protease and
Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Inc.).
After centrifugation at 15,000 RCF for 30 min at 4°C, supernatants
were recovered and used for immunoblot analysis. The proteins were
separated by SDS-PAGE and then transferred to polyvinylidene
difluoride (PVDF) membranes (Merck Millipore, Darmstadt, Germany).
The blots were blocked with 5% BSA in TBST buffer (Thermo Fisher
Scientific, Inc.) and then probed with antibodies against total
AMPK (1:1,000), phosphor-AMPK T172 (1:1,000) and GAPDH overnight at
4°C. After washing, the blots were incubated with HRP-conjugated
secondary antibodies at room temperature for 1 h and visualized by
super ECL detection reagent (Applygen, Beijing, China).
Statistical analysis
Statistics was evaluated using GraphPad Prism 5.0
software (GraphPad Software, Inc.). The unpaired Student's t-test
was used for the data analysis, in addition to analysis of variance
plus a post-hoc test. All data were shown as mean ± standard error
(SE). P<0.05 was considered to indicate a statistically
significant difference.
Results
AMPK is activated in radiation
resistant colon cancer patients
A previous study reported that ionizing radiation
induced time- and dose-dependent phosphorylation of AMPK at Thr172
(12). To investigate whether the
activation of AMPK is clinically correlated with resistance to
radiation, the phosphorylation statuses of AMPK and total AMPK were
assessed in radioresistant colon cancer patient samples. Notably,
phosphorylated AMPK and total AMPK were upregulated in
radioresistant patients and radiosensitive patients (Fig. 1A; P<0.001), indicating that AMPK
may contribute to radiation resistance in colon cancer. In
addition, RT-qPCR results demonstrated that the mRNA levels of AMPK
were upregulated in radioresistant cancer samples compared with
radiosensitive patient samples (Fig.
1B; P<0.001). These results suggest there is a correlation
between AMPK and radioresistance, and therefore targeting AMPK may
be a potential therapeutic strategy for overcoming tumor radiation
resistance.
Radiation resistant colon cancer cells
display elevated AMPK activity and expression
To further examine the roles of AMPK in radiation
resistance, a radiation resistant colon cancer cell line was
generated from DLD-1 parental cells by exposure to repeated
irradiation for 2 months. Radioresistant cell clones were selected
and pooled for the following experiments. To verify the
radioresistance, parental cells (sensitive) and resistant pool
cells were irradiated with different doses from 0–16 Gy for 24 h.
Cell viability assays demonstrated that DLD-1 radioresistant cells
tolerated higher doses of radiation compared with radiosensitive
cells. The IC50 of the resistant cells was 20 Gy, which
was ~10-fold greater than the IC50 of the radiosensitive
cells, which exhibited significant inhibition of viability at 1–16
Gy (Fig. 2A). The phosphorylation of
AMPK and total AMPK were upregulated in DLD-1 radioresistant cells
(Fig. 2B) in addition to the mRNA
level of AMPK (Fig. 2C; P<0.01),
consistent with the above results that AMPK pathway was upregulated
in radiation resistant colon cancers.
Overexpression of AMPK in colon cancer
cells contributes to radiation resistance
To address whether AMPK activation is involved in
the radiosensitivity of colon cancer cells, the cell survival
profiles of DLD-1 cells was analyzed in cells that overexpressed
AMPK. AMPK was successfully upregulated in cells transfected with
the overexpression vector compared with the control (Fig. 3A); overexpression in colon cancer
cells led to increased resistance to radiation treatments (Fig. 3B; P<0.01), indicating activation of
AMPK pathway contributes to radioresistance. To investigate
further, DLD-1 cells were treated with metformin, which is a
well-studied stimulator of AMPK pathway, at the indicated
concentrations. Metformin alone (0.5–2.5 µM) induced a
dose-dependent activation of AMPK, it also significantly enhanced
the activation of AMPK under radiation treatment (Fig. 4C; P<0.01 with 2.5 µM metformin).
The effects of irradiation and metformin on DLD-1 cells survival.
The surviving fraction of metformin pretreated-cells following
radiation treatments were significantly higher compared with
non-treated cells (Fig. 4D;
P<0.01, following 10 Gy IR and 2 µM metformin versus 0 µM
metformin), indicating that the activation of the AMPK pathway may
have contributed to colon cancer cells resistance to radiation.
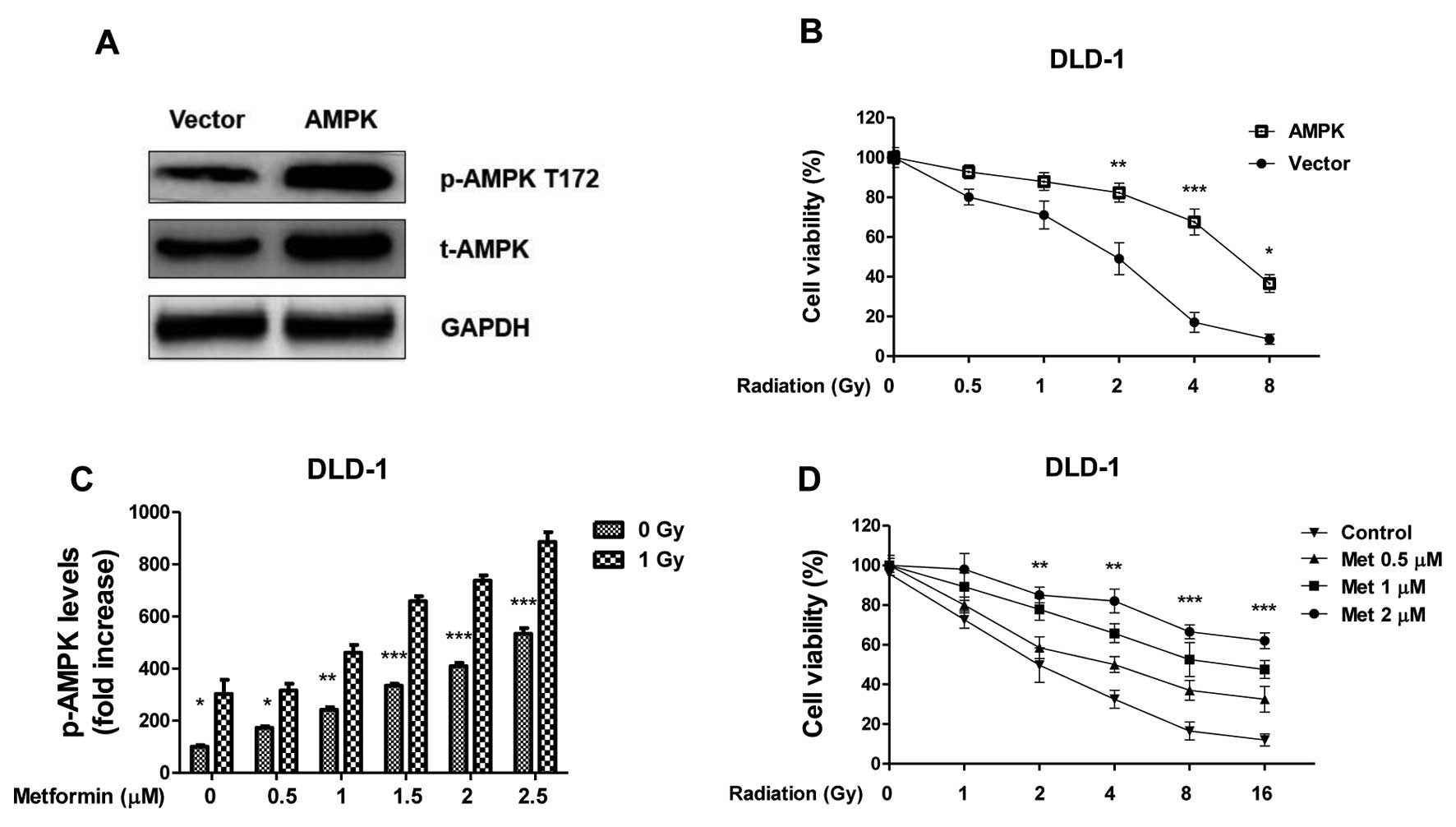 | Figure 3.Overexpression of AMPK renders renal
cancer cells resistance to radiation. (A) DLD-1 cells were
transfected with AMPK or control vector for 48 h, the expression of
total AMPK and phospho-AMPK at T172 were measured by western blot
analysis. GAPDH was a loading control. (B) DLD-1 cells were
transfected with AMPK or control vector for 48 h, followed by
irradiation at 0, 0.5, 1, 2, 4 and 8 Gy. Then cells were analyzed
by cell viability assay. (C) DLD-1 cells were treated with
Metformin at the indicated concentrations for 1 h, then the
phospho-AMPK at T172 were measured by western blot, the relative
intensities were showed as folds increase. (D) DLD-1 cells were
treated with Metformin at the indicated concentrations for 1 h,
then cells were exposed to radiation at 0, 1, 2, 4, 8 and 16 Gy,
followed by the measurements of cell viability. Columns, mean of
three independent experiments; bars, standard error. *P<0.05 vs.
control group; **P<0.01 vs. control group; ***P<0.001 vs.
control group. AMPK, adenosine monophosphate-activated kinase;
t-AMPK, total AMPK; p-AMPK, phospho-AMPK; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; T172, phosphorylation
site on Threonine 172. |
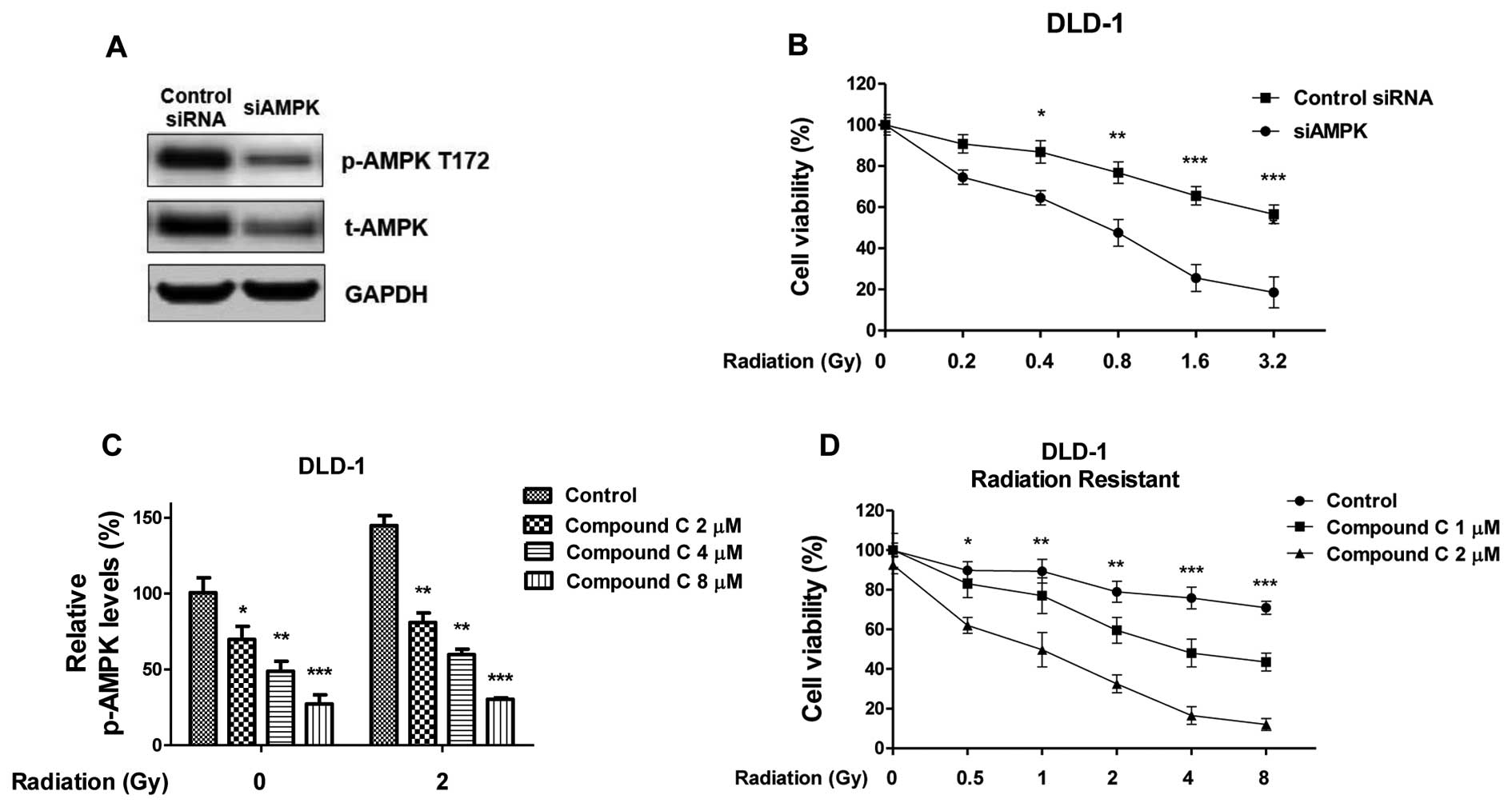 | Figure 4.Inhibition of AMPK re-sensitizes
radiation resistant cells. (A) DLD-1 radiation resistant cells were
transfected siAMPK or control siRNA for 48 h, the expression of
t-AMPK and p-AMPK at T172 were measured by western blot analysis.
GAPDH was a loading control. (B) Cells with siAMPK or control siRNA
were treated with radiation at indicated doses, followed by cell
viability assay. (C) Inhibition of the activity of AMPK by Compound
C. DLD-1 radiation resistant cells were treated with Compound C at
the indicated concentrations for 1 h, then the p-AMPK at T172 were
measured by western blot analysis, the relative intensities are
presented as fold increase. (D) DLD-1 radiation resistant cells
were treated with Compound C at the indicated concentrations for 1
h, then cells were treated with radiation at indicated doses,
followed by cell viability assay. Columns, mean of three
independent experiments; bars, standard error. *P<0.05;
**P<0.01; ***P<0.001. siRNA, small interfering RNA; AMPK,
adenosine monophosphate-activated kinase; t-AMPK, total AMPK;
p-AMPK, phospho-AMPK; GAPDH, glyceraldehyde-3-phosphate
dehydrogenase. |
Inhibition of AMPK re-sensitizes
radiation-resistant colon cancer cells
To investigate whether targeting the AMPK pathway
could attenuate radiation resistance, molecular and biochemical
inhibition of AMPK was achieved by knocking down expression of AMPK
by siRNA and treatment with Compound C (CC) which is an inhibitor
of AMPK pathway (Fig. 4A). In DLD-1
cells, knocking down of AMPK by anti-AMPK α subunit siRNA led to a
significant suppression of cell survival rates following radiation
(Fig. 4B), consistent with the above
results that DLD-1 cells became resistant to radiation with the
overexpression of AMPK (Fig. 3B). CC
treatment inhibited the activities of AMPK (Fig. 4C) and the inhibition of AMPK with CC
(2, 4 and 8 µM) abolished the radiation-activated of AMPK,
suggesting that blocking the AMPK pathway may be effective in
resensitizing radioresistant cancer cells to radiation. To evaluate
the effects of treating radioresistant colon cancer cells with the
combination of AMPK inhibitor and radiation, DLD-1 radiation
resistant cells were treated with CC (1 and 2 µM CC). The cells
were then treated with radiation at the indicated dosages. CC
treatment in radiation resistant colon cancer cells significantly
promoted the susceptibility to radiation at multiple doses and the
AMPK pathway was significantly inhibited (Fig. 4D). Taken together, the results
suggested that the combination of AMPK inhibitor and radiation
showed a synergistic inhibitory effect on radioresistant colon
cancer cells.
Discussion
A previous study reported that ionic radiation
increased AMPK protein levels in lung cancer and breast cancer
cells (12). In the present study,
the importance of AMPK in radiation resistance in colon cancer
cells was assessed and highlighted the clinical relevance by
inhibition of AMPK, suggesting a therapeutic role of AMPK inhibitor
for overcoming radiation resistance. Furthermore, RT-qPCR analysis
demonstrated an induction of AMPK mRNA expression in radiation
resistant cells, suggesting that the AMPK expression is regulated
at the translational and transcriptional level. However, the
detailed mechanisms for the downstream regulator of AMPK remain to
be elucidated. A preliminary study suggested that IR stimulates
AMPKb1/2 gene expression in HCT116 and H1299 cells in a
p53-dependent manner (11): Putative
p53 consensus binding sites were identified on the AMPKb1 and b2
promoters. The upstream regulators of AMPK in response to radiation
therefore require further study.
Metformin is a widely used drug for treatment of
type 2 diabetes and it activates AMPK leading to the induction of
fatty acid oxidation and glucose uptake (13). In addition, it has been reported that
chronic activation of AMPK may also induce the expression of muscle
hexokinase and glucose transporters (Glut4) (14). The present study indicates that the
activation of AMPK by metformin contributes to radiation resistance
in colon cancer cells, suggesting that AMPK-mediated
radioresistance may be affected by cancer metabolic regulators.
Notably, inhibition of AMPK by CC re-sensitized radiation resistant
cells. AMPK has been reported to promote glycolysis of cancer cells
as an adaptive response to metabolic stress (15). The present study provides evidence
that the activated AMPK contributes to radioresistance in colon
cancer cells. Since chemo- and radio-resistant cancer cells exhibit
elevated levels of glycolysis, the present results indicates that
AMPK-mediated radioresistance may be a result of up-regulation of
glycolysis in cancer cells. Future studies should investigate the
molecular mechanisms of this phenotype by measuring the glycolysis
ratio of radioresistant cells and determining the signal pathway
that regulates AMPK-mTOR-Akt in response to radiation. In
conclusion, the present study indicated the important roles of AMPK
pathway in radiation resistance in cancer cell lines and colon
cancer patient samples and highlights a novel strategy for the
development of therapeutic agents to reverse radiation
resistance.
Acknowledgements
The authors would like to thank the staff and
faculty members working in the Department of General Surgery, China
Japan Union Hospital of Jilin University and Dr. Zhuo Liu for
editorial assistance.
References
|
1
|
Rastogi RP Richa, Kumar A, Tyagi MB and
Sinha RP: Molecular mechanisms of ultraviolet radiation-induced DNA
damage and repair. J Nucleic Acids. 2010:5929802010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pajonk F, Vlashi E and McBride WH:
Radiation resistance of cancer stem cells: The 4 R's of
radiobiology revisited. Stem Cells. 28:639–648. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galanty Y, Belotserkovskaya R, Coates J,
et al: Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses
to DNA double-strand breaks. Nature. 462:935–939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lavin MF: Ataxia-telangiectasia: From a
rare disorder to a paradigm for cell signalling and cancer. Nat Rev
Mol Cell Biol. 9:759–769. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shen C and Houghton PJ: The mTOR pathway
negatively controls ATM by up-regulating miRNAs. Proc Natl Acad Sci
USA. 110:11869–11874. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gurumurthy S, Xie SZ, Alagesan B, et al:
The Lkb1 metabolic sensor maintains haematopoietic stem cell
survival. Nature. 468:659–663. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hardie DG: AMPK: Positive and negative
regulation, and its role in whole-body energy homeostasis. Curr
Opin Cell Biol. 33:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jones RG, Plas DR, Kubek S, et al:
AMP-activated protein kinase induces a p53-dependent metabolic
checkpoint. Mol Cell. 18:283–293. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Inoki K, Kim J and Guan KL: AMPK and mTOR
in cellular energy homeostasis and drug targets. Annu Rev Pharmacol
Toxicol. 52:381–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sanli T, Storozhuk Y, Linher-Melville K,
et al: Ionizing radiation regulates the expression of AMP-activated
protein kinase (AMPK) in epithelial cancer cells: Modulation of
cellular signals regulating cell cycle and survival. Radiother
Oncol. 102:459–465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sanli T, Rashid A, Liu C, et al: Ionizing
radiation activates AMP-activated kinase (AMPK): A target for
radiosensitization of human cancer cells. Int J Radiat Oncol Biol
Phys. 78:221–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rena G, Pearson ER and Sakamoto K:
Molecular mechanism of action of metformin: Old or new insights?
Diabetologia. 56:1898–1906. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Habegger KM, Hoffman NJ, Ridenour CM, et
al: AMPK enhances insulin-stimulated GLUT4 regulation via lowering
membrane cholesterol. Endocrinology. 153:2130–2141. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu SB and Wei YH: AMPK-mediated increase
of glycolysis as an adaptive response to oxidative stress in human
cells: Implication of the cell survival in mitochondrial diseases.
Biochim Biophys Acta. 1822:233–247. 2012. View Article : Google Scholar : PubMed/NCBI
|
















