Introduction
von Hippel-Lindau (VHL) disease is an autosomal
dominantly inherited neoplastic syndrome that increases
susceptibility to a variety of benign and malignant neoplasms,
including retinal and central nervous system (CNS)
hemangioblastomas, clear cell renal cell carcinomas (RCCs),
pheochromocytomas, pancreatic neuroendocrine tumors (PanNETs) and
endolymphatic sac tumors (1). In
addition, renal cysts, pancreatic cysts or cystadenomas, and
epididymal or broad ligament cystadenomas may also occur (2).
The identification of the VHL gene has allowed for
the development of molecular genetic diagnosis of VHL disease by
direct mutation analysis (3). It has
been well documented that VHL patients may develop further de
novo lesions, occasionally many years after the initial
diagnosis, despite the complete excision of initial neoplasm
(4). Thus, long-term follow-ups for
patients with VHL disease are necessary. Clinical courses are
occasionally complicated, and are of interest because VHL involves
multiple organs but results in organ-specific tumors (5). The current study reports the case of a
patient with VHL disease complicated with lateral ventricular
hemangioblastomas and, subsequently, PanNET, bilateral renal cysts
and clear cell RCC.
Case report
A 17-year-old male presented to The Second
Affiliated Hospital of Zhejiang University School of Medicine
(Hangzhou, China) in September 2007, with a progressive headache
and dizziness of three months duration. He had experienced no
previous medical problems except for a single episode of
generalized convulsion 2 months prior to admission. The
neurological examination was normal. Magnetic resonance imaging
(MRI) of the brain was performed using a 1.5-T MR imaging system
(Signa; GE Healthcare, Milwaukee, WI, USA) and revealed a mass near
the septum pellucidum, and a subependymal nodule at a same level in
the right lateral ventricle. The mass near the septum pellucidum
was hypointense on T1-weighted images and hyper-isointense on
T2-weighted images relative to the normal white matter.
Gadolinium-enhanced T1-weighted MRI demonstrated a solid, rounded,
intraventricular tumor of 2 cm in diameter, with strong contrast
enhancement. However, the subependymal nodule was hyper-isointense
on T1-weighted images and hypointense on T2-weighted images, which
indicated hemorrhage. The nodule exhibited slight contrast
enhancement (Fig. 1A–D).
In September 2007, the patient underwent a
right-sided parietal craniotomy. The mass and subependymal nodule
were completely excised via a trans-sulcal approach through the
superior parietal lobule. Grossly, the mass was cherry-red,
foam-like and highly vascular. However, the subependymal nodule was
brownish, slightly firm and was adherent to the ventricular
ependyma. The resected specimen was fixed with 4% neutral
formaldehyde, followed by conventional dehydration, paraffin
embedding, sectioning, and hematoxylin and eosin (HE) staining.
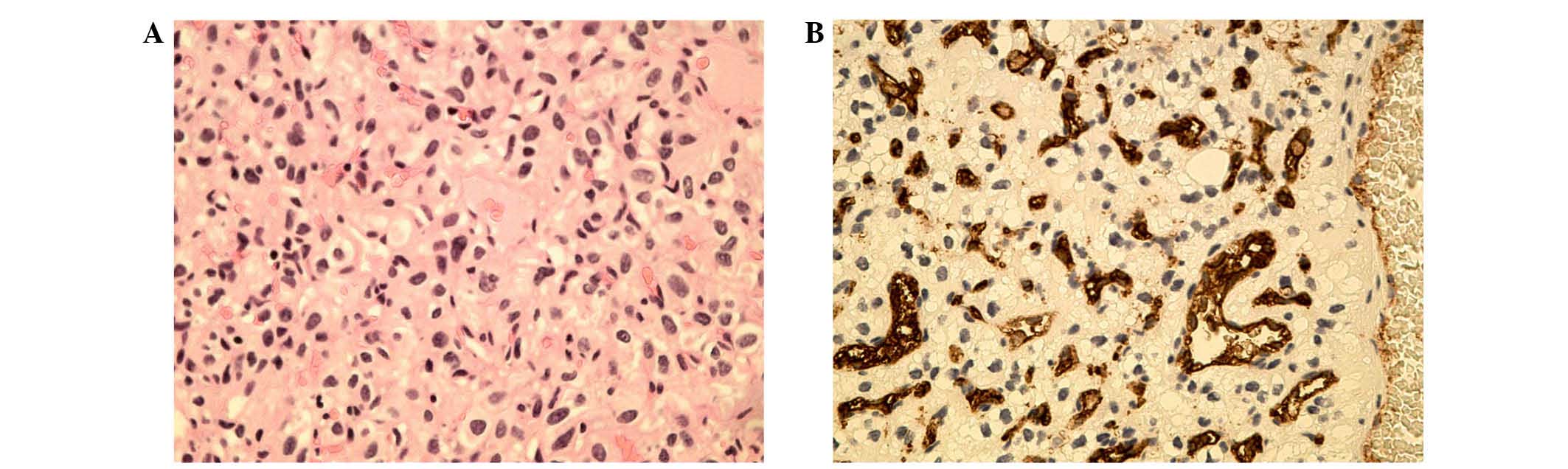
Histopathological examination of both lesions revealed tissue
composed of many small blood vessels separated by numerous
polygonal stromal cells, with lightly stained cytoplasm.
Additionally, hemorrhagic foci were observed within the tissue of
the subependymal nodule, in accordance with the MRI findings
(Fig. 1). Immunohistochemical
staining was performed using the EnVision two-step method.
Antibodies to CD34 (mouse monoclonal 1:100 dilution), chromogranin
A (mouse monoclonal 1:100 dilution), synaptophysin (mouse
monoclonal 1:50 dilution) and Ki67 (mouse monoclonal 1:50
dilution), which were all purchased from Funakoshi, Co., Ltd.
(Tokyo, Japan). Immunohistochemical staining revealed strong
immunopositivity in several stromal cells for vimentin, epithelial
membrane antigen and neuron specific enolase. The intercapillary
component was immunopositive for the endothelial cell marker CD34.
Histopathological examination and immunohistochemical staining
confirmed that the two lesions were typical hemangioblastomas
(Fig. 2A–B) (6).
Postoperatively, the patient experienced a transient
sensory aphasia, which recovered fully within 2 weeks; the
subsequent course was uneventful. According to clinical criteria
(7), a suspected diagnosis of VHL
disease was established for this patient. Thus, additional
systematic enquiry and examinations associated with VHL disease
were performed, including funduscopic examination, abdominal spiral
computed tomography (CT; LightSpeed VCT 64; GE Healthcare,
Pittsburgh, PA, USA), magnetic resonance (MR) scanning of the
spinal cord, and 24-h urinary analysis for vanillylmandelic acid,
metanephrines and total catecholamines. The results of the
monitoring workup were negative, with the exception of multiple
bilateral renal cysts that were visualized by abdominal CT
examination. No family history of specific diseases was reported. A
mutation analysis was completed; however, following polymerase
chain reaction and sequencing of three exons of the VHL gene, no
variation was detected.
At 10 pm on 12th December 2013, the patient was
readmitted to the emergency department of The Second Affiliated
Hospital of Zhejiang University School of Medicine due to an attack
of syncope and loss of consciousness without convulsion of the
limbs. Emergency laboratory data confirmed hypoglycemic shock
(plasma glucose level, 1.18 mmol/l; normal fasting glucose range,
3.89–6.1 mmol/l). The patient's symptoms were alleviated
significantly following the administration of glucose via
intravenous injection. Subsequently, pancreatic exploration was
conducted, and pancreatic routine ultrasonography suggested a
hypoechoic lesion of 3 cm in diameter on the distal body of the
pancreas. Contrast-enhanced ultrasonography with enhancement
material, consisting of SonoVue (Bracco, Milan, Italy) diluted with
5 ml of normal sodium, was performed and revealed a pancreatic
solid tumor with marked contrast enhancement (Fig. 3A). During ultrasound examination
(SSA-790A; Toshiba Medical Systems, Tokyo, Japan) with a convex
probe (PVT-375BT; 3.75-MHz center frequency), multiple small cysts
and multiple small hypoechoic solid lesions were detected in the
bilateral kidneys. Thus, abdominal CT and MRI were performed to
further characterize these complex lesions. Unenhanced and enhanced
abdominal CT scans failed to delineate the pancreatic lesion
clearly, except for a focal swelling and contour-deformation of the
distal body of the pancreas (Fig.
3B). However, all of the pancreatic and renal lesions (cystic
and solid) were delineated clearly using a Signa HDx 3.0T MR
scanner (GE Healthcare, Milwaukee, WI, USA). The pancreatic lesion
was hyperintense relative to normal pancreatic tissue on
fat-suppressed T2-weighted sequences, and hypointense on
fat-suppressed unenhanced T1-weighted sequences (Fig. 3C). Additional diffusion-weighted
imaging (DWI) was also acquired using a single-shot,
echo-planar-imaging sequence with short tau inversion
recovery-based fat suppression (Fig.
3D). Water diffusion was measured at b-values of 0 and 1,000
sec/mm2. A homogeneous, well-defined mass of 3 cm in
diameter, which had high signal intensity and was located in the
distal body of the pancreas, was delineated more clearly on DWI
(b=1,000 sec/mm2), and there were decreased apparent
diffusion coefficients (ADCs) on the ADC map as compared with the
adjacent normal tissue of the pancreas. Regions of interest (ROI)
were drawn within the tumor and the normal-appearing pancreas. The
measured ADC values were 0.610×10−3 mm2/sec
in the tumor and 1.143×10−3 mm2/sec in the
normal-appearing pancreas. On dynamic gadolinium-based
contrast-enhanced and fat-suppressed T1-weighted images, the lesion
exhibited early enhancement during the arterial phase and
progressive fill-in and enhancement during the delayed phase.
In accordance with the ultrasonographic findings,
abdominal CT imaging with contrast revealed multiple bilateral
cystic and solid renal lesions of varying sizes (range, 2–20 mm).
The solid lesions had marked contrast enhancement, whereas the
cystic lesions did not (Fig. 4A).
Similarly, renal MRI revealed multiple cystic lesions and multiple
solid lesions in both kidneys. The multiple cystic lesions (8 in
the right kidney, 2–10 mm in diameter; and 6 in the left kidney,
2–8 mm in diameter) exhibited homogeneous hyperintensity on
T2-weighted images and hypointensity on fat-suppressed T1-weighted
images, and no enhancement on contrast-enhanced and fat-suppressed
T1-weighted images. The solid lesions (4 in the right kidney, 3–20
mm in diameter; and 3 in the left kidney, 2–10 mm in diameter) were
hypointense on T1- and T2-weighted images and had strong
enhancement on contrast-enhanced and fat-suppressed T1-weighted
images. DWI with a b-value of 1,000 sec/mm2 revealed
that the signal intensities were reduced for all cystic lesions and
increased considerably for all solid lesions (Fig. 4B). ADC maps confirmed these findings,
yielding high values (0.97×10−3 mm2/sec) for
all cystic lesions and low values (0.57×10−3
mm2/sec) for all solid lesions. These data suggested a
preoperative diagnosis of PanNET and multiple bilateral renal cysts
and RCCs.
On 7th January 2014, due to the malignant potential
of the PanNET and the multifocal spread of the bilateral renal
tumors, simultaneous distal splenopancreatectomy and enucleation of
the bilateral renal tumors were performed. A lesion of 3 cm in
diameter within the distal body of the pancreas was identified. The
tumor was a well-demarcated, red-brown mass, and its cut surface
appeared solid and homogeneous. At histological examination, the
tumor was characterized as a well-differentiated PanNET (8); immunohistochemically the tumor exhibited
chromogranin A and synaptophysin positivity, and a Ki-67 index
<2% (Fig. 5A–C).
Intraoperatively, 6 tumors in the right kidney and 5
tumors in left kidney could be reached and were enucleated. The
renal vein and inferior vena cava were normal, with no obvious
abnormal lymph nodes. Histopathological examination revealed
typical clear cell RCC (pT1, cN0, cM0, G2, R0), which contained
clear cells with inconspicuous nucleoli forming prominent
microcysts (9), in both kidneys
(Fig. 5D).
The patient's postoperative course was uneventful
and he was discharged on the 10th postoperative day, with a regular
follow-up recommended. At follow-up to date, the patient is
asymptomatic and investigations have revealed no tumor recurrence
or presence of any further masses in any other organ. Informed
consent was obtained from the patient. The study was approved by
the patient and the Second Affiliated Hospital, School of Medicine,
Zhejiang University Service Ethics Committee (Zhejiang, China).
Discussion
Hemangioblastomas of the CNS are uncommon,
accounting for ~2% of primary CNS tumors (1,3). These
tumors may occur sporadically; however, they have been found to be
associated with VHL disease in ~30% of cases. Hemangioblastomas are
predominantly detected in the cerebellum, spinal cord or brainstem
(in ~83% of cases, collectively) (2).
Less common locations include the leptomeninges and sellar-sphenoid
sinus (4,7,10).
Supratentorial locations are rare, representing 4% of sporadic
hemangioblastomas and 13% of those associated with VHL disease
(10). Supratentorial
intraventricular hemangioblastomas are extremely rare, and only 7
cases (excluding the present case) of lateral ventricular
hemangioblastomas have been reported in the English literature to
date; 6 were identified to be associated with VHL disease and 1
case was undefined. Thus, the strong possibility of VHL disease
should be raised in patients with hemangioblastomas in such unusual
locations (10).
In the current case, establishing a preoperative
diagnosis of hemangioblastoma of the lateral ventricle was
extremely challenging based on imaging findings. The lesions were
suspected to be ‘subependymal astrocytoma’ based on the patient's
age and certain imaging features, such as the location of the
lesion and the coexistent tumor and subependymal nodule. According
to clinical criteria (7), patients
with ≥2 CNS hemangioblastomas or 1 CNS hemangioblastoma and a
VHL-associated visceral tumor fulfill the criteria for clinical
diagnosis without a familial history of VHL disease (7,11,12). Thus, a final diagnosis of VHL disease
could be established in the current patient due to the 2
hemangioblastomas and the multiple bilateral cysts.
With respect to the natural history of VHL disease,
Wanebo et al (13) reported
that VHL-associated tumors have two separate phases of growth,
termed rapid and quiescent growth periods, if the patient is
observed for a sufficient duration. A rapid growth period is
typically followed by a quiescent period that can last >30
months. In the present case, 75 months after the initial surgery,
the PanNET was identified. Furthermore, the cystic masses in the
kidneys subsequently developed into RCCs.
Pancreatic tumors or cysts develop in 35–77% of VHL
patients in the majority of case series, with most of the cysts
being benign (14). Pancreatic tumors
include cystadenomas (12%), hemangioblastomas (<1%),
adenocarcinomas (<1%) and PanNETs (9–17%) (15). PanNETs are tumors with an abundant
blood supply, and the pancreas is an organ with high perfusion.
Therefore, on contrast CT scanning, pancreatic tumors and the organ
itself exhibit simultaneously enhancement. The difference in
density is not as obvious as for pancreatic carcinoma, which has a
poor blood supply. Large PanNETs sometimes appear only as a change
in the contour of the pancreas, as in the current case, whilst
small PanNETs are easily missed at diagnosis. MRI with superior
tissue contrast is able to show PanNETs better, particularly on DWI
sequences when detecting small lesions. Thus, MRI (and most
importantly DWI) is essential for monitoring the pancreas in VHL
patients.
PanNETs associated with VHL disease are typically
nonfunctional and are located throughout the pancreas. These tumors
are often asymptomatic; however, they can cause pancreatitis or a
mass-related pain (16). Patients can
occasionally present hypoglycemic symptoms, such as in the present
case, due to neoplastic expansion or local invasion. This would be
an indication for surgical treatment. Resection is recommended for
tumors >3 cm, and the patients should be followed up closely
(17).
VHL-associated pancreatic lesions are always
associated with renal lesions (18).
Patients with VHL disease are at high risk of developing multiple
renal cysts and RCCs, which occurs in around two-thirds of patients
(19). The most frequent histological
form of RCC is clear cell carcinoma which, in the majority of
cases, tends to be multifocal and involve both kidneys. Although
renal cysts may be benign, they are considered premalignant
lesions; RCCs may arise from cystic or non-cystic renal parenchyma
(20). The primary therapeutic
approach is surgery, aiming to preserve the maximal amount of renal
parenchyma (21). The onset of renal
tumors represents the predominant cause of mortality in patients
with VHL syndrome (~70%); thus, early diagnosis followed by a
possible partial nephrectomy in order to avoid dialysis and renal
transplantation is extremely important (21,22).
In conclusion, the current case suggests that a high
degree of suspicion for VHL disease should be raised in patients
with hemangioblastomas in supratentorial locations, particularly
when intraventricular. In addition, follow-up of VHL patients must
not be discontinued after their initial diagnosis and treatment,
even if they experience symptom-free periods. Furthermore, MR
examination, particularly DWI sequences, is valuable when abdominal
organs, such as the pancreas or kidneys, are involved. In summary,
the current study emphasizes the importance of close follow-ups
with radiological examination of CNS hemangioblastoma and other
organs subsequent to the initial presentation of CNS
hemangioblastoma associated with VHL disease.
References
|
1
|
Maher ER and Kaelin WG Jr: Von
Hippel-Lindau disease. Medicine (Baltimore). 76:381–391. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaelin WG: Von Hippel-Lindau disease. Annu
Rev Pathol. 2:145–173. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Latif F, Tory K, Gnarra J, Yao M, Duh FM,
Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, et al:
Identification of the von Hippel-Lindau disease tumor suppressor
gene. Science. 260:1317–1320. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kaelin WG Jr: Molecular basis of the VHL
hereditary cancer syndrome. Nat Rev Cancer. 2:673–682. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gossage L, Eisen T and Maher ER: VHL, the
story of a tumour suppressor gene. Nat Rev Cancer. 15:55–64. 2014.
View Article : Google Scholar
|
|
6
|
Sundaram C, Rammurti S, Reddy JJ, Prasad
SS and Purohit AK: Hemangioblastoma: A study of radiopathologic
correlation. Neurol India. 51:373–375. 2003.PubMed/NCBI
|
|
7
|
Lonser RR, Glenn GM, Walther M, Chew EY,
Libutti SK, Linehan WM and Oldfield EH: Von Hippel-Lindau disease.
Lancet. 361:2059–2067. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Batcher E, Madaj P and Gianoukakis AG:
Pancreatic neuroendocrine tumors. Endocr Res. 36:35–43. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee C, Park JW, Suh JH, Nam KH, et al:
Histologic variations and immunohistochemical features of
metastatic clear cell renal cell carcinoma. Korean J Pathol.
47:426–432. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takeuchi S and Takasato Y: Supratentorial
intraventricular hemangioblastomas. Acta Neurol Belg. 111:353–356.
2011.PubMed/NCBI
|
|
11
|
Johnston LB, Chew SL, Lowe D, Reznek R,
Monson JP and Savage MO: Investigating familial endocrine neoplasia
syndromes in children. Horm Res. 55(Suppl 1): S31–S35. 2001.
View Article : Google Scholar
|
|
12
|
Gaal J and de Krijger RR: Neuroendocrine
tumors and tumor syndromes in childhood. Pediatr Dev Pathol.
13:427–441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wanebo JE, Lonser RR, Glenn GM and
Oldfield EH: The natural history of hemangioblastomas of the
central nervous system in patients with von Hippel-Lindau disease.
J Neurosurg. 98:82–94. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tamura K, Nishimori I, Ito T, et al:
Diagnosis and management of pancreatic neuroendocrine tumor in von
Hippel-Lindau disease. World J Gastroenterol. 16:4515–4518. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hammel PR, Vilgrain V, Terris B, Penfornis
A, Sauvanet A, Correas JM, Chauveau D, Balian A, Beigelman C,
O'Toole D, et al: Pancreatic involvement in von Hippel-Lindau
disease. The Groupe Francophone d'Etude de la Maladie de von
Hippel-Lindau. Gastroenterology. 119:1087–1095. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chetty R, Kennedy M, Ezzat S and Asa SL:
Pancreatic endocrine pathology in von Hippel-Lindau disease: An
expanding spectrum of lesions. Endocr Pathol. 15:141–148. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Blansfield JA, Choyke L, Morita SY, Choyke
PL, Pingpank JF, Alexander HR, Seidel G, Shutack Y, Yuldasheva N,
Eugeni M, et al: Clinical, genetic and radiographic analysis of 108
patients with von Hippel-Lindau disease (VHL manifested by
pancreatic neuroendocrine neoplasms (PNETs). Surgery. 142:814–818;
discussion 818.e1–e2. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mukhopadhyay B, Sahdev A, Monson JP,
Besser GM, Reznek RH and Chew SL: Pancreatic lesions in von
Hippel-Lindau disease. Clin Endocrinol (Oxf). 57:603–608. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chetty R, Kennedy M, Ezzat S and Asa SL:
Pancreatic endocrine pathology in von Hippel-Lindau disease: An
expanding spectrum of lesions. Endocr Pathol. 15:141–148. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nickerson ML, Jaeger E, Shi Y, Durocher
JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko
V, et al: Improved identification of von Hippel-Lindau gene
alterations in clear cell renal tumors. Clin Cancer Res.
14:4726–4734. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patard JJ, Shvarts O, Lam JS, Pantuck AJ,
Kim HL, Ficarra V, Cindolo L, Han KR, De La Taille A, Tostain J, et
al: Safety and efficacy of partial nephrectomy for all T1 tumors
based on an international multicenter experience. J Urol.
171:2181–2185; quiz 2435. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bratslavsky G, Liu JJ, Johnson AD,
Sudarshan S, Choyke PL, Linehan WM and Pinto PA: Salvage partial
nephrectomy for hereditary renal cancer: Feasibility and outcomes.
J Urol. 179:67–70. 2008. View Article : Google Scholar : PubMed/NCBI
|