Introduction
Heavy chain diseases (HCDs) are a group of rare,
systemic syndromes, generally associated with variants of B cell
neoplasms, and are characterized by the production of an abnormal
immunoglobulin (Ig) heavy chain that is incapable of binding to
light chains. HCDs are named according to the type of heavy chain
produced: IgA (α-HCD), IgG (γ-HCD) and IgM (µ-HCD). The incidence
of γ-HCD is higher than that of α- but lower than that of µ-HCD
(1). The median age at diagnosis is
68 years (range, 42–87 years). Patients with HCDs may present with
a large number of constitutional symptoms and may exhibit
concomitant autoimmune disease (2).
The clinical course of γ-HCD is extremely variable and ranges from
an asymptomatic benign, or stable process to a rapidly progressive
neoplasm leading to mortality within a few weeks. γ-HCD generally
presents as a lymphoproliferative disorder, comprising
lymphadenopathies, splenomegaly and constitutional symptoms. Due to
the heterogeneity of γ-HCD, the choice of therapy should entirely
rely on the underlying disorder and clinicopathologic features.
Prognosis is variable and the mean survival time has been reported
to be 7.4 years (range, 1 month to >21 years) (3,4). Rare as B
cell disorders are, it is even rarer for patients to exhibit
HCD-associated T cell disorders, and only a few cases have been
reported to date (2,5,6). The aim
of the present study was to report a case of γ-HCD presenting with
T cell receptor (TCR) gene rearrangements, in order to provide
additional biological insight into the nature of γ-HCD.
Case report
An 81-year-old man presented with enlargement of the
left submandibular lymph nodes in August 2012 at the Cancer
Hospital of the Chinese Academy of Medical Sciences (Beijing,
China). The patient had been diagnosed with testicular sarcoma and
had undergone orchiectomy 12 years prior to admission. His past
medical history additionally included hypertension, coronary
atherosclerotic heart disease and coronary stent implantation.
Physical and ultrasound examination revealed enlarged submandibular
lymph nodes. Computed tomography (CT; Ingenuity Core 128; Philips
Medical Systems, Inc., Bothell, WA, USA) scanning confirmed
multiple enlarged lymph nodes involving the mediastinum, axilla,
diaphragmatic, retroperitoneum, and common iliac, external iliac
and inguinal lymph nodes. Deep cervical lymph node biopsy suggested
hyperplastic lymphoid tissue, primarily in the T region, along with
local neutrophil infiltration. In addition, the biopsy indicated
that the cell population was polyclonal. Immunohistochemical
staining revealed cluster of differentiation (CD)19+,
CD20+, CD21+ (normally atrophied follicular
dendritic cells only), CD23+ (normally follicular
dendritic cells), CD2+, CD3+,
CD5+, B cell lymphoma (Bcl)-2++,
Bcl-6−, cyclin D1 (CCND1)−, Ki67+
(10%) and scattered large cells expressing CD30. The following
monoclonal primary antibodies were used for immunohistochemistry
(all purchased from OriGene Technologies; ZSGB-BIO, Beijing,
China): Mouse anti-human CD19 (cat. no. TA506236; 1:100); mouse
anti-human CD20 (cat. no. TA800394; 1:150) rabbit anti-human CD21
(cat. no. TA327627; 1:100); rabbit anti-human CD23 (cat. no.
TA506412; 1:150); mouse anti-human CD2 (cat. no. TA500394; 1:100);
mouse anti-human CD3 (cat. no. TA506064; 1:100); mouse anti-human
CD5 (cat. no. TA501335; 1:100); mouse anti-human CD30 (cat. no.
TA801630; 1:100); mouse anti-human Bcl-2 (cat. no. TA803003;
1:150); mouse anti-human Bcl-6 (cat. no. TA804186; 1:150); rabbit
anti-human cyclin D1 (cat. no. ZA-0101; 1:100); and mouse
anti-human Ki67 (cat. no. TA802736; 1:100). Horseradish peroxidase
conjugated goat anti-mouse and anti-rabbit monoclonal IgG (cat. no.
PV-6000; ZSGB-BIO) were used as the secondary antibodies. The
PV-9000 kit containing the reagents used for immunohistochemistry
was purchased from OriGene Technologies (ZSGB-BIO), and tissue
samples were prepared (including staining and sectioning) according
to the manufacturer's protocol. PCR analysis (Applied Biosystems™
GeneAmp™ PCR System 9700; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) was performed using DNA obtained from the patient's bone
marrow and IdentiClone™ IGH/IGK/IGL/TCRB/TCRD/TCRG Gene Clonality
Assays (Invivoscribe Technologies, Inc., San Diego, CA, USA)
according to the manufacturer's protocol. The PCR cycling
conditions were as follow: initial denaturation at 95°C for 7 min,
followed by 35 cycles of 95°C for 45 sec, 65°C for 45 sec and 72°C
for 90 sec, followed by a final step at 72°C for 10 min. This
revealed clonal TCR gene rearrangements, positive for TCRβ-B,
TCRβ-C and TCRγ-A. Based on these findings, a diagnosis of
malignant lymphoma involving the tonsil, nasopharyngeal top wall
and right side of the pericardium was suspected. The patient was
discharged in September 2012, and subsequently, a chemotherapy
regimen of cyclophosphamide (100 mg/24 h), etoposide (100 mg/24 h)
and prednisone (30 mg/24 h) was administered for 12 months.
Repeated CT scans revealed that the size of the lymph nodes had
markedly reduced with treatment.
In December 2013, the patient presented with dyspnea
and was referred to the Department of Pneumology (Luhe Hospital,
Capital Medical University, Beijing). Upon physical examination,
the patient was observed to have an enlarged spleen, as well as
bilateral cervical and axillary lymphadenopathies. CT scans of the
chest, abdomen and pelvis confirmed splenomegaly and bilateral
axillary, hilus pulmonis, mediastinal and retroperitoneal lymph
node enlargement, accompanied by polyserous effusions, including
bilateral pleural, pericardial, abdominal and pelvic cavity
effusions. Thoracocentesis results suggested transudate pleural
effusion, which was additionally analyzed by flow cytometry (FCM;
FACSCalibur; BD Biosciences, Franklin Lakes, NJ). FCM demonstrated
primarily T cells positive for CD38, CD7, CD5, CD4, CD3, CD2 and
CD3, and weakly positive for CD10. The
CD4+CD3+/CD8+CD3+ ratio
was 3.62. The following reagents (all purchased from BD
Biosciences) were used for flow cytometry: BD Simultest™ Anti-human
κ fluorescein isothiocyanate (FITC)/λ phycoerythrin (PE; dilution,
1:10); monoclonal mouse anti-human allophycocyanin (APC)-labeled
CD19 antibody (catalog no., 340437; dilution, 1:40); monoclonal
mouse anti-human FITC-labeled CD138 antibody (catalog no., 347191;
dilution, 1:10); monoclonal mouse anti-human APC-labeled CD38
(HB-7) antibody (catalog no., 345807; dilution, 1:40); monoclonal
mouse anti-human APC-labeled immunoglobulin (Ig)G1
(catalog no., 340442; dilution, 1:40); monoclonal mouse anti-human
PE-labeled IgG1 (catalog no., 349043; dilution, 1:10);
and monoclonal mouse anti-human FITC-labeled IgG1
(catalog no., 349041; dilution, 1:10). All samples and reagents
were prepared according to the manufacturer's protocol.
Routine laboratory tests revealed leukopenia [white
blood cells, 1.61×109/l (normal range,
4–10×109/l); neutrophils, 92.11% (normal range, 45–77%);
lymphocytes, 5.13% (normal range, 20–40%)] accompanied by anemia
[hemoglobin, 68 g/l (normal range, 120–160 g/l)] and slight
thrombocytopenia [platelet count, 97×109/l (normal
range, 100–300×109/l)], with a normal peripheral blood
smear. The erythrocyte sedimentation rate was observed to be
increased [25 mm/h (normal range, 0–20 mm/h)]. Total protein plasma
concentration was 61.5 g/l (normal range, 64–83 g/l), with albumin
levels that were lower than normal [25.2 g/l (normal range, 38–53
g/l)] and globulin levels of 36.2 g/l. Blood urea nitrogen and
creatinine levels were within the normal ranges. Skeletal X-rays
(DigitalDiagnost Pro; Philips Medical Systems, Inc.) did not reveal
any bone lesions.
The patient was subsequently transferred to the
Department of Hematology (Luhe Hospital, Capital Medical
University, Beijing) with the clinical suspicion of a
lymphoproliferative disorder, and more specific tests were
performed. Bone marrow aspirate revealed 10% of bone marrow nuclear
cells were atypical lymphoplasmacytic cells. Bone marrow FCM
confirmed 5.08% of bone marrow nuclear cells were atypical
population of lymphoplasmacytic cells, which were CD19+,
CD56−, CD38+, CD138+, cytoplasmic
IgG− and cytoplasmic λ+ (Fig. 1). PCR analysis revealed abnormal
clonal Igκ rearrangement, positivity for Vκ-kappa deleting element
(Kde) and INTR-Kde, and negativity for Ig heavy locus (IGH) forward
1–3 primers, Vλ-Jλ, DH1-6-JH and DH7-JH. TCRα/β gene rearrangements
were observed. The TCRγ gene demonstrated no rearrangements, while
the TCRδ gene was weakly positive in the rearrangement assay.
Karyotyping revealed a normal male karyotype. Fluorescence in
situ hybridization testing did not detect any abnormalities in
the copy numbers of IGH, 1q21 (a commonly used probe in the
prognosis of plasmacyte diseases), tumor protein p53 and
retinoblastoma 1, or any IGH-v-maf avian musculoaponeurotic
fibrosarcoma oncogene homolog, fibroblast growth factor receptor
3-IGH, IGH-CCND1 and D13S319 (a probe that detects abnormalities at
13q14.3 in chromosomes) fusion genes.
Serum protein electrophoresis (HYDRASYS 2; Sebia
Ltd., Camberley, UK) revealed an increased β region, along with
decreased albumin levels and γ regions. Furthermore, a loss of
separation between the β1 and β2 regions, caused by a narrow spike,
was observed (Fig. 2A), A 41.0%
(normal range, 60.3–71.4%) 2.52 g/dl; α1 3.8% (normal range,
1.4–2.9%) 0.23 g/dl; α2 1.5% (normal range, 7.2–11.3%) 0.09 g/dl; β
+M 51.3% (normal range, 8.1–12.7%) 3.15 g/dl; γ 2.4% (normal range,
8.7–16.0%) 0.15 g/dl. Immunoglobulin quantification revealed an
increased level of IgG (74.4 g/l; normal range, 7–16 g/l),
accompanied by decreased levels of IgA (0.49 g/l; normal range,
0.7–4 g/l), IgM (0.004 g/l; normal range, 0.4–2.3 g/l), C3 (0.752
g/l; normal range, 0.9–1.8 g/l) and C4 (0.053 g/l; normal range,
0.1–0.4 g/l). Serum immunofixation electrophoresis (IFE; HYDRASYS
2; Sebia Ltd., Camberley, UK) revealed the presence of a monoclonal
band in the γ-heavy chain lane without the corresponding band for
the light chain lanes (Fig. 2B).
Serum free light chains included both decreased free κ (115 mg/dl)
and λ (79.6 mg/dl) chains, while the κ/λ ratio was normal (1.44).
Urine IFE revealed the presence of an unusual spike in the β region
(Fig. 3A), with an increased urine
IgG concentration of 0.31 g/dl (total IgG, ~4.65 g/24 h), and a
small amount of associated λ light chain and protein-free λ light
chain (Fig. 3B); however, the IgG
subclasses were all lower than normal [IgG1, <0.43 mg/l (normal
range, 4900–11400 mg/l); IgG2, <84.4 mg/l (normal range,
1500–6400 mg/l); IgG3, 75 mg/l (normal range, 200–1100 mg/l); IgG4,
41 mg/l (reference interval, 80–1400 mg/l)].
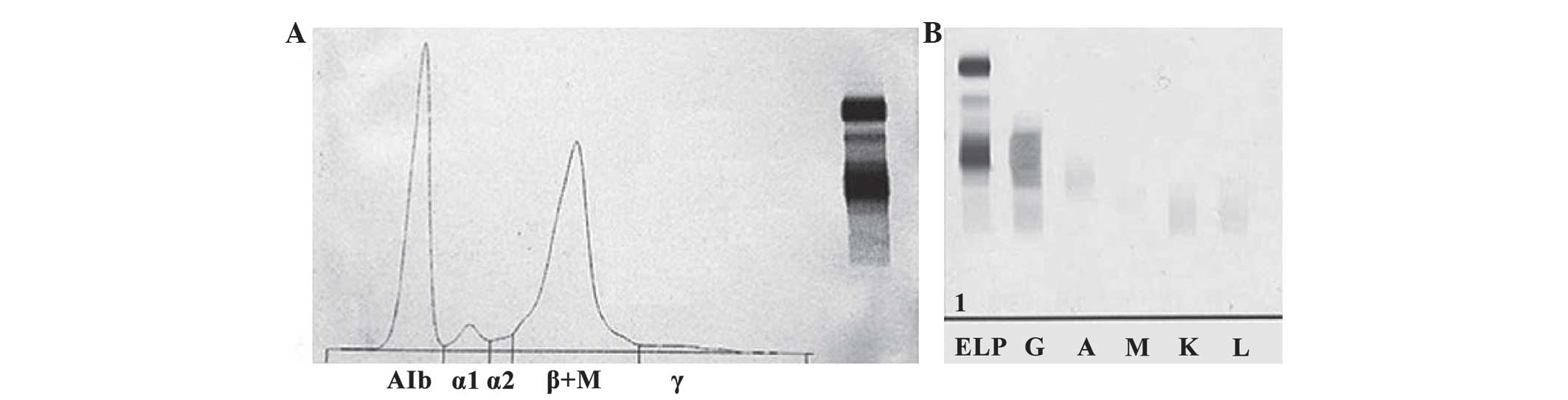 | Figure 2.SPEP and IFE. (A) SPEP showing an
increased β region, along with decreased albumin levels and γ
regions, as well as a loss of separation between the β1 and β2
regions caused by a narrow spike. (B) Serum IFE showing the
presence of a monoclonal band in the γ-heavy chain lane without the
corresponding band for the light chain lanes. SPEP, serum protein
electrophoresis; IFE, immunofixation electrophoresis; ELP,
electrophoresis of serum; G, gamma heavy chains; A, albumin; M, µ
light chains; K, κ light chains; L,.λ light chains. |
Based on the aforementioned laboratory findings, the
patient was diagnosed with γ-HCD. Dyspnea was treated by thoracic
injection of 10 mg dexamethasone, which relieved the symptom to a
marked extent. Repeated X-rays revealed that the pleural effusions
had disappeared; however, 1 month later, the patient developed
acute pancreatitis with increased levels of serum lipase [768 µ/l
(normal range, 23–300 µ/l)] and amylase [1,249 µ/l (normal range,
30–110 µ/l)]. As a result, the patient succumbed to persistent high
fever and pancytopenia on Feb 25, 2014.
Discussion
Heavy chain diseases (HCDs) are rare B cell
lymphoplasma cell proliferative disorders that are characterized by
the production of incomplete monoclonal immunoglobulin heavy chains
without the associated light chains (3). As the clinical manifestations of HCDs
lack specificity, the presence of HCD proteins (a group of abnormal
immunoglobulin heavy chains incapable of binding to light chains)
is considered the only diagnostic criterion of the disease
(1).
HCDs have three main Ig subtypes: IgG, IgM and IgA.
Among them, γ-HCD (IgG subtype) is the rarest, with only ~150 cases
reported in the literature to date (7). The classification of the
lymphoplasmacytic disorder underlying γ-HCD has been controversial
(2,4,7,8). The most common subclass of γ-HCD is
IgG1, which accounts for 65% of all γ-HCD cases. Other subclasses
include IgG3 (27% of all γ-HCD cases), IgG4 (3% of all γ-HCD cases)
and IgG2 (2% of all γ-HCD cases) (2).
It has been reported that the majority of γ-HCD cases resemble
lymphoplasmacytic lymphoma; however, γ-HCD has been reported to be
associated with a wide variety of disorders (2,4). In a
similar manner to cases of multiple myeloma, γ-HCD cases
demonstrated elevated IgG levels corresponding to depressed IgM and
IgA (9). The clinical features of
γ-HCD include fever, anemia, lymph node enlargement, hepatomegaly,
splenomegaly, and in some cases slight leukopenia and
thrombocytopenia (2,7). However, the course and prognosis of
γ-HCD may vary widely depending on the heterogeneity of the
clinicopathological features of the disease (10).
With regard to the present patient, the serum IFE
confirmed a monoclonal γ globulin band with no corresponding light
chain, therefore suggesting γ-HCD. However, the patient's serum
levels of IgG subtypes 1–4 were demonstrated to be lower than
normal, which had not been previously reported in the literature to
the best of our knowledge (11). As
the summation of the IgG subtypes (<2.4 g/l) was markedly lower
compared with the total IgG level (74.4 g/l), an antigen excess
(also known as excess high-dose hook effect) caused by
nephelometric assays was considered to be a potential explanation,
as this may have resulted in false negative data (12–14). The
discrepancies between the values may additionally have been due to
a failure of the IgG subclass antigen to recognize the lost
antigenic domain in incomplete heavy chain components. Another
discrepancy was observed between the globulin (36.2 g/l) and total
immunoglobulin values (74.4 g/l). This could be attributed to the
lack of specificity of the biuret test used to detect the protein.
In addition, the decreased levels of C3, C4, IgA and IgM may
indicate inadequate protein synthesis, leading to infiltration of
the immune system in advanced disease.
In addition, both lymph node biopsy and bone marrow
aspirate revealed TCR gene rearrangement in the present patient.
The bone aspirate additionally revealed Igκ gene rearrangement.
Both TCR gene rearrangement in B cell lymphoma (7), and Igκ gene rearrangement in γ-HCD
(2) have been reported in the
literature; however, to the best of our knowledge, TCR gene
arrangement in γ-HCD has not yet been reported. In the present
case, the exudative nature of the pleural effusion and
immunophenotyping of CD4− expressing T cell subset
indicated strong antigen stimulation or immune dysregulation, which
may result in CD4+T lymphocyte activation. Putative
extrinsic or intrinsic antigens may trigger B and T cell activation
and proliferation, resulting in gene rearrangements in
immunoglobulin heavy chains and/or TCR gene clusters (6). As a result of gene deletion of the
γ-heavy chain locus, structurally defective IgGs may be formed,
leading to γ-HCD (6).
To the best of our knowledge, the present study
reports the first case of TCR gene rearrangement in γ-HCD. The
present study revealed an alternative manifestation of γ-HCD, which
may provide additional biological insights into this rare B cell
disorder.
References
|
1.
|
Bianchi G, Anderson KC, Harris NL and
Sohani AR: The heavy chain diseases: Clinical and pathologic
features. Oncology (Williston Park). 28:45–53. 2014.PubMed/NCBI
|
|
2.
|
Fermand JP, Brouet JC, Danon F and
Seligmann M: Gamma heavy chain “disease”: Heterogeneity of the
clinicopathologic features. Report of 16 cases and review of the
literature. Medicine (Baltimore). 68:321–335. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Franklin EC, Lowenstein J, Bigelow B and
Meltzer M: Heavy chain disease - a new disorder of serum
gamma-globulins: Report of the first case. Am J Med. 37:332–350.
1964. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Wahner-Roedler DL, Witzig TE, Loehrer LL
and Kyle RA: Gamma-heavy chain disease: Review of 23 cases.
Medicine (Baltimore). 82:236–250. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Okuda K, Himeno Y, Toyama T, Ohta M,
Kitagawa M and Sugai S: Gamma heavy chain disease and giant lymph
node hyperplasia in a patient with impaired T cell function. Jpn J
Med. 21:109–114. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Zhang L, Sotomayor EM, Papenhausen PR,
Shao H, Moscinski LC, Sandin RL, Caceres G, Valenica H, Malafa M,
List AF and Sokol L: Unusual concurrence of T-cell large granular
lymphocytic leukemia with Franklin disease (gamma heavy chain
disease) manifested with massive splenomegaly. Leuk Lymphoma.
54:205–208. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Bieliauskas S, Tubbs RR, Bacon CM, Eshoa
C, Foucar K, Gibson SE, Kroft SH, Sohani AR, Swerdlow SH and Cook
JR: Gamma heavy-chain disease: Defining the spectrum of associated
lymphoproliferative disorders through analysis of 13 cases. Am J
Surg Pathol. 36:534–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Swerdlow SH, Campo E, Harris NL, Jaffe ES,
Pileri SA, Stein H, Thiele J and Vardiman JW: WHO Classification of
Tumours of Haematopoietic and Lymphoid Tissues. 2:(4th). Lyon,
France: IARC Press. 194–199. 2008.
|
|
9.
|
Ogawa K, Imai M, Tanigawa T, Tsuji T and
Arima T: Pathological studies on a long-term survived case of gamma
heavy chain disease - a brief review of 30 reported cases and a
proposal for histological typing. Acta Pathol Jpn. 28:759–778.
1978.PubMed/NCBI
|
|
10.
|
Arnason JE and Mendez LM: Induction
therapy for gamma-heavy chain disease with bortezomib and
dexamethasone: A case report. Blood. 120:50512012.
|
|
11.
|
Wahner-Roedler DL and Kyle RA: Heavy chain
diseases. Best Pract Res Clin Haematol. 18:729–746. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Daval S, Tridon A, Mazeron N, Ristori JM
and Evrard B: Risk of antigen excess in serum free light chain
measurements. Clin Chem. 53:1985–1986. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Johannis W, Blommer J, Klatt AR, Renno JH
and Wielckens K: Gamma heavy chain disease in a patient with
rheumatoid arthritis - a laboratory evaluation. Biochem Med
(Zagreb). 22:373–379. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Busser B, Millet S, Bulabois CE, Faure P
and Renversez JC: Unusual increased beta-globulins in an elderly
patient. Clin Chem. 57:948–951. 2011. View Article : Google Scholar : PubMed/NCBI
|
















