Introduction
Acute myeloid leukemia (AML) is the most common type
of leukemia in adults, and anthracycline-based chemotherapy is the
principal option for the treatment of AML. Despite advances in AML
treatment, 20% of AML patients do not respond to induction
chemotherapy, and 40–60% of patients relapse (1). The majority of therapeutic failures are
due to cellular resistance to anti-leukemic drugs (2). Therefore, it is important to identify
strategies that reverse resistance to drugs.
The development of refractory AML remains unclear,
and may involve various molecular mechanisms, including the altered
expression of one or several multidrug resistance (MDR) genes,
including multidrug resistance-associated protein 1 (MRP1), lung
resistance related protein (LRP) and P-glycoprotein (P-gp)
(3,4).
Previous studies have investigated altered glucose metabolism in
chemoresistant leukemia cells (5–14). It has
been reported that prednisolone resistance in patients with
precursor B-cell acute lymphoblastic leukemia (ALL) was associated
with an increased expression of genes involved in glucose
metabolism (5,6). Imatinib-resistant chronic myelogenous
leukemia cells also exhibited a high glycolysis phenotype, with
elevated glucose uptake and lactate production compared with
imatinib-sensitive cells (7–10). In addition, AML cell lines with an
increased level of glycolysis showed increased resistance to the
induction of apoptosis in vitro due to a combination of
all-trans retinoic acid and arsenic trioxide (11). Furthermore, inhibition of glycolysis
by glycolytic inhibitors, such as 2-deoxy-D-glucose (2-DG),
lonidamine (LND) and 3-bromopyruvate (3BrPA), or downregulation of
the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
by RNA interference sensitizes prednisolone-resistant ALL cell
lines to glucocorticoids (12). Acute
leukemia subtypes, including pre-B-cell ALL, T-cell ALL and AML,
demonstrated growth arrest and cell death when treated with the
novel glycolysis inhibitor 3-BrOP. Potentiated adenosine
triphosphate (ATP) depletion and pro-apoptotic effects were
observed subsequent to treatment with 3-BrOP. Combined with the
cytochrome c-reductase inhibitor antimycin A and the
mammalian target of rapamycin (mTOR) inhibitor rapamycin, treatment
with 3-BrOP resulted in effects potentiated by ATP-depletion and
pro-apoptotic effects in these leukemia cells (13). In several human cancer cell lines,
including the Jurkat, HeLa and U937 cell lines, glycolysis
inhibition dramatically enhances apoptosis induced by Fas or tumor
necrosis factor-related apoptosis-inducing ligand. This
sensitization is controlled through adenosine
monophosphate-activated protein kinase, which is the central
energy-sensing system of the cell (14). Although this study supported that
leukemia cells with high glucose metabolism were associated with
chemoresistance, it remains unknown whether glycolysis-associated
genes are involved in the MDR of AML.
Glycolysis is considered to be the main source of
energy for tumor cells, and is much less efficient at producing
energy compared with oxidative phosphorylation (15). The process of glycolysis produces only
2 mol ATP per 1 mol glucose, while oxidative phosphorylation of
glucose results in ~36 mol ATP per 1 mol glucose. Each reaction in
the glycolytic pathway is catalyzed by a specific glucose
transporter (GLUT) or enzyme, such as GLUT1, hexokinase (HK),
phosphofructokinase (PFK), GAPDH and enolase (ENO) (16,17). Tumor
cells shift their metabolism from oxidative phosphorylation towards
the less efficient glycolysis, independent of the presence of
oxygen. This metabolic change is associated with factors including
the upregulation of glycolytic enzymes and glucose transporters,
decreased expression of oxidative phosphorylation enzymes and a
lower mitochondrial content (18).
The present study investigated the glycolytic
activity and expression of associated molecules in drug-resistant
AML cells in comparison with drug-sensitive AML cells. The
increased expression of glycolytic pathway-associated genes,
including hypoxia inducible factor (HIF)-1α, HK-II, GLUT1, and
lactate dehydrogenase (LDH), and low expression of β-F1-ATPase were
shown to be associated with drug resistance in AML cells. It was
also observed that inhibition of glycolysis through the use of
glycolytic inhibitors rendered resistant AML cells susceptible to
Adriamycin (ADR). The present data suggested that
glycolysis-associated genes are involved in AML drug resistance and
may be promising therapeutic targets for AML therapy.
Patients and methods
Patient samples
Bone marrow and serum samples were obtained from 90
newly diagnosed and 18 relapsed patients with AML in The First
Affiliated Hospital of Jishou University (Jishou, Hunan, China)
between October 2010 and November 2011. Patients with AML M3 were
excluded. All samples contained ≥80% leukemia cells. Within 24 h of
sampling, mononuclear cells from bone marrow samples were obtained
subsequent to red blood cell lysis (Wuhan Boster Biological
Technology, Ltd., Wuhan, Hubei, China) of the lower fraction
subsequent to density gradient centrifugation at 480 × g. Isolated
mononuclear cells were washed twice by phosphate-buffered saline
(PBS; Wuhan Boster Biological Technology, Ltd.), and stored in
liquid nitrogen until use. The present study was performed in
accordance with the modified Declaration of Helsinki (19), and the protocol was approved by The
Medicine Ethics Committee of Zhongshan City People's Hospital
(Zhongshan, Guangdong, China) prior to the start of the study.
Informed consent was obtained from all patients and healthy
volunteers.
Cell lines and cell culture
The human AML HL-60 cell line and the ADR-resistant
HL-60/ADR sub-line were obtained from the Laboratory of Hematology
of Nanfang Hospital of South Medical University (Guangzhou, China).
These cell lines were cultured at 37°C in a 5% humidified
atmosphere in Gibco RPMI-1640 medium (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% HyClone fetal bovine
serum (FBS; GE Healthcare Life Sciences, Logan, UT, USA), 100
units/ml penicillin (Guangzhou Ucando Biotechnology Co., Ltd.,
Guangzhou, China), 100 µg/ml streptomycin (Guangzhou Ucando
Biotechnology Co., Ltd.)and 2 mM glutamine (Guangzhou Ucando
Biotechnology Co., Ltd.). Prior to each experiment, HL-60/ADR cells
were treated with 1 µmol/ml ADR (Sigma-Aldrich, St. Louis, MO, USA)
for 10–28 days and then cultured for 10 days without ADR exposure
(20).
Glucose consumption assay
Glucose consumption was detected by the conversion
of glucose to 6-phosphogluconate and reduced nicotinamide adenine
dinucleotide (NADH) with the Glucose (HK) Assay kit
(Sigma-Aldrich), according to the manufacturer's protocol. Briefly,
106 HL-60 and HL-60/ADR cells were cultured in RPMI-1640
containing 2 g/l glucose. Following 4 days of culture, the medium
was collected by centrifugation at 715 × g to remove the cells, and
incubated at room temperature for 2 h with glucose assay buffer
(Sigma-Aldrich), containing 1.5 mM NAD, 1 mM ATP, 1 U/ml hexokinase
and 1 U/ml glucose-6-phosphate dehydrogenase. During this time,
glucose was phosphorylated to glucose-6-phosphate.
Glucose-6-phosphate was then oxidized to 6-phospho-gluconate in the
presence of oxidized nicotinamide adenine dinucleotide (NAD),
reducing NAD to an equimolar amount of NADH. The conversion of NAD
to NADH was measured by the increase in absorbance at 340 nm using
a spectrophotometer (UV-1800; Shimadzu, Kyoto, Japan), which was
directly proportional to the glucose concentration.
RNA extraction and quantitative
polymerase chain reaction (qPCR)
Total RNA from cell lines and primary mononuclear
cell samples were extracted using Invitrogen TRIzol reagent (Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
Subsequent to removal of the DNase I, cDNA was reverse transcribed
from 1 µg of total RNA in 20 µl reaction volume, in accordance with
the maufacturer's instructions for the PrimeScript® RT
kit (Takara Biotechnology Co., Ltd., Dalian, Liaoning, China). The
PCR machine was ABI7500 (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The sequences of the primers were as follows:
HIF-1α forward, 5′-ACAGCCTCACCAAACAGAGCAGG-3′ and reverse,
5′-CGCTTTCTCTGAGCATTCTGCAAAGC-3′; GLUT1 forward,
5′-GCGGGTTGTGCCATACTCATGACC-3′ and reverse,
5′-AGGCCACAAAGCCAAAGATGGCC-3′; and β-actin forward,
5′-TGGCACCCAGCACAATGAA-3′ and reverse,
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′. qPCR was performed using a SYBR
Green reaction kit (Takara Biotechnology Co., Ltd.) and an Applied
Biosystems PRISM 7500 real-time PCR detection system (Thermo Fisher
Scientific, Inc.). The expression levels of HIF-1α and GLUT1 were
analyzed relative to the levels of the β-actin gene transcript.
Sequences of the primers (Shanghai GenePharma Co., Ltd., Shanghai,
China) were shown in Table I. The
reaction mixture for each direct qPCR was performed using 10 µl
SYBR Premix Ex (Takara Biotechnology Co., Ltd.), 0.8 µl (0.4 µM) of
each primer and 2 µl cDNA, and in a total reaction volume of 20 µl.
The qPCR conditions consisted of pre-denaturation at 95°C for 30
sec, followed by 40 cycles of denaturation at 95°C for 3 sec and
annealing at 60°C for 30 sec. Three independent experiments were
performed on the cells in independent cultures at 3 different
times.
 | Table I.Primer sequences used in reverse
transcription-qunatitative polymerase chain reaction. |
Table I.
Primer sequences used in reverse
transcription-qunatitative polymerase chain reaction.
| Primer | Sequence | Product size,
bp |
|---|
| β-actin |
|
|
|
Sense |
5′-TGGCACCCAGCACAATGAA-3′ | 187 |
|
Antisense |
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′ |
|
| HIF-1α |
|
|
|
Sense |
5′-ACAGCCTCACCAAACAGAGCAGG-3′ | 163 |
|
Antisense |
5′-CGCTTTCTCTGAGCATTCTGCAAAGC-3′ |
|
| GLUT1 |
|
|
|
Sense |
5′-GCGGGTTGTGCCATACTCATGACC-3′ | 99 |
|
Antisense |
5′-AGGCCACAAAGCCAAAGATGGCC-3′ |
|
| HK-II |
|
|
|
Sense |
5′-TCTCCGCCTCGGTTTCCCAACT-3′ | 154 |
|
Antisense |
5′-AAGAGGGTCTCATCAGAGAGGCGC-3′ |
|
Western blot analysis
Cells (2×106) were washed twice in
ice-cold PBS and then incubated for 10 min on ice with 300 µl lysis
buffer, which consisted of 1% Triton X-100, 0.1% SDS, 50 mM Tris
(pH 8.0), 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 0.1 mM
Na3VO4, 0.1 mM benzamidine, 5 µg/ml leupeptin
and 5 µg/ml aprotinin (Sigma-Aldrich). Whole-cell lysates were
clarified by centrifugation at 21,130 × g for 15 min at 4°C. The
protein concentration was determined using the Bradford protein
assay (Bio-Rad Laboratories, Hercules, CA, USA) with bovine serum
albumin (Bio-Rad Laboratories) as a standard protein. In total, 30
µg of protein was separated by electrophoresis on a sodium dodecyl
sulfate-polyacrylamide gel (10% gel; Sigma-Aldrich). Subsequent to
electrophoresis, proteins were transferred to a nitrocellulose
membrane (Millipore, Billerica, MA, USA). Following 1 h incubation
in a blocking solution containing 5% non-fat dry milk in PBS and
0.5% Tween-20 (Sigma-Aldrich) the membrane was blotted with the
mouse monoclonal anti-β-F1-ATPase antibody (catalog no., sc-55597;
dilution, 1:500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
and mouse monoclonal anti-β-actin (control) antibody (catalog no.,
sc-47778; dilution, 1:1,000; Santa Cruz Biotechnology, Inc.)
overnight at 4°C. Subsequent to washing, the blot was incubated
with the secondary antibodies (dilution, 1:1,000; Santa Cruz
Biotechnology, Inc.) for 1 h at room temperature. The
immunoreactive bands were visualized with the enhanced
chemiluminescence (ECL) reagent (Pierce Biotechnology, Inc.,
Rockford, IL, USA), according to the manufacturer's protocol. All
the tests were repeated three times.
Enzyme-linked immunosorbent assay
(ELISA)
Patient serum levels of LDH were measured using
Quantikine kit (R&D Systems, Inc., Minneapolis, MN, USA),
according to the manufacturer's instructions. Optical density was
measured using an ELX800 ELISA reader (Bio-Tek, Winooski, VT, USA)
at 492 mm. The results were calculated from a standard curve
generated by dilutions of a known amount of recombinant LDH
protein. The LDH concentration in each sample was the average of 3
independent experiment results.
Methyl thiazolyl tetrazolium (MTT)
assay
The MTT assay was performed to assess the effect of
3BrPA or 2-DG on the proliferation of AML cell lines and
responsiveness to ADR. The final drug concentrations of ADR ranged
between 0.25 and 5 µg/ml in HL-60/ADR cells and between 0.001 and
0.1 µg/ml in HL-60 cells. The half maximal inhibitory concentration
(IC50), which is defined as the drug concentration at
which cell growth is inhibited by 50%, was used as a measure of
cellular drug resistance. Various concentrations of ADR (0.25–5
µg/ml for HL-60/ADR cells; 0.001–0.1 µg/ml for HL-60 cells) were
used due to differences in ADR resistance, but corresponded with
the induction of similar amounts of cell death. Inhibition of
glycolysis was established by the addition of 0.25–2.00 mM 2-DG
(Sigma-Aldrich) or 20 µM 3BrPA (Sigma-Aldrich). HL-60 or HL-60/ADR
cells were plated in 96-well plates at a density of
5×104 cells/well for the proliferation assay. Subsequent
to 48 h, the cells were stained with 10 µl sterile MTT dye (5
mg/ml; Sigma-Aldrich) at 37°C for 4 h, and the culture medium was
then removed. Subsequently, 200 µl dimethyl sulfoxide
(Sigma-Aldrich) was added to the plates and thoroughly combined for
10 min. Spectrometric absorbance at 570 nm was measured using the
ELx800 Absorbance Microplate Reader (Bio-Tek). All groups were
assessed 6 times.
Coefficient of drug interaction
(CDI)
The CDI is used to analyze the synergistic
inhibitory effect of drug combinations. CDI is calculated as
follows: CDI = AB / (A × B), where AB is the ratio of absorbance of
the combination groups to the control group and A or B is the ratio
of the absorbance of the single agent group to the control group.
Thus, a CDI of <1, 1 or >1 indicates that the drugs are
synergistic, additive or antagonistic, respectively. A CDI <0.7
indicates that the drugs are significantly synergistic (21).
Flow cytometry
Annexin V-fluorescein isothiocyanate (FITC)
apoptosis detection kit (KeyGen, Nanjing, Jiangsu, China) was used
for the detection of apoptotic cells, according to the
manufacturer's instructions. Briefly, 1–5×105 HL-60 or
HL-60/ADR cells were collected and washed with PBS. From the cell
suspension, cells were re-suspended in 1X binding buffer and then
incubated with 5 µl Annexin V-FITC and 5 µl propidium iodide
solution and incubated for 10 min in the dark. Fluorescence of the
cells was determined immediately using flow cytometry with
CellQuest 3.2 software (Becton Dickinson, San Jose, CA, USA).
Staining with Annexin V and 10 µl PI was performed according to the
manufacturer's protocol for the Annexin V-FITC Apoptosis Detection
kit (Sigma-Aldrich). Cells in the early stages of apoptosis stain
with Annexin V-FITC conjugate alone.
Statistical analysis
All statistical analysis was performed using the
statistical software package SPSS 16.0 (SPSS, Inc., Chicago, IL,
USA). Student's t-test was used to determine the
significance of the differences between mean values. One-way
analysis of variance was performed and post-hoc multiple
comparisons were performed using the least significant difference
test under homogeneity of variance, while a comparison of the means
of multi-group samples was performed using Dunnett's T3 method
under homogeneity of variance. The results were expressed as the
mean ± standard error, and P<0.05 was considered to indicate a
statistically significant difference.
Results
Patient characteristics
Using the chemotherapy response criteria established
by the Chinese Chemotherapy Symposium of Leukemia in 1987 (22), chemotherapy response was assessed
according to two induction cycles of chemotherapy. The response
criteria consisted of complete remission (CR), partial remission
(PR) and no remission (NR). CR was defined as ≤5% blasts in
normocellular or hypercellular bone marrow with a normal peripheral
count (granulocyte count, >1×109/l; platelet count,
>1×1011/l). PR required similar criteria, with the
exception of the presence of 6–20% marrow blasts. NR was defined as
failure to achieve CR or PR. The clinical characteristics of the
three patient groups are shown in Table
II. In addition, 38 healthy donors acted as a negative
control.
| Table II.Clinical characteristics of patients
with acute myeloid leukemia that demonstrated different
sensitivities to chemotherapy. |
Table II.
Clinical characteristics of patients
with acute myeloid leukemia that demonstrated different
sensitivities to chemotherapy.
|
| CR | PR | NR |
|
|---|
|
|
|
|
|
|
|---|
|
Characteristics | Mean | n (%) | Mean | n (%) | Mean | n (%) | P-value |
|---|
| Total |
| 64 (59.3) |
| 16 (14.8) |
| 28 (25.9) |
|
| Age | 43.2 |
| 44.6 |
| 36.3 |
| NS |
| <60
years |
| 52 (81.3) |
| 14 (87.5) |
| 26 (92.9) |
|
| ≥60
years |
| 12 (18.8) |
| 2
(12.5) |
| 2 (7.1) |
|
| Gender |
|
|
|
|
|
| NS |
|
Male |
| 18 (56.3) |
| 10 (62.5) |
| 16 (57.1) |
|
|
Female |
| 14 (43.8) |
| 6
(37.5) |
| 12 (42.9) |
|
| Blasts, % | 86.9 |
| 87.0 |
| 89.5 |
| NS |
| Disease status |
|
|
|
|
|
| NS |
| Newly
diagnosed |
| 58 (90.6) |
| 14 (87.5) |
| 18 (64.3) |
|
|
Relapsed |
| 6 (9.4) |
| 2
(12.5) |
| 10 (35.7) |
|
| MICM |
|
|
|
|
|
| NS |
| M2 |
| 42 (65.6) |
| 14 (87.5) |
| 14 (50.0) |
|
| Others
(M4, M5, M7) |
| 22 (34.4) |
| 2
(12.5) |
| 4
(50.0) |
Glycolysis in AML cell lines
The glucose consumption in ADR-resistant HL-60/ADR
cells following 4 days of incubation was set as 100%. The
ADR-resistant AML HL-60/ADR cell line showed increased glucose
consumption (97.4±0.70%) compared with the ADR-sensitive HL-60 cell
line (55.0±3.16%) subsequent to 4 days of culture (P<0.001).
HIF-1α, GLUT1 and HK-II messenger
(m)RNA expression in primary AML cells and cell lines
As shown in Fig. 1,
the transcription levels of the HIF-1α, GLUT1 and HK-II genes,
which are involved in glucose metabolism, showed increased
expression in the healthy controls, CR group, PR group and NR group
(P<0.001). The level of HIF-1α mRNA in the NR group (14.85±4.16)
was evidently increased compared with the healthy controls
(0.79±0.22; P<0.001), CR group (1.67±0.55; P<0.001) and PR
group (4.02±1.10; P<0.001). GLUT1 mRNA expression was also
markedly higher in the NR group (25.17±11.89) compared with the
healthy controls (0.34±0.11; P<0.001), CR group (0.83±0.24,
P<0.001) and PR group (4.34±1.35; P<0.001). Similarly, the
expression of HK-II mRNA was significantly upregulated in the NR
group (17.47±5.22) compared with the healthy controls (0.49±0.20;
P<0.001), CR group (0.78±0.39; P<0.001) and PR group
(11.61±3.69; P=0.036).
Subsequently, the expression levels of these three
genes in the AML ADR-sensitive HL-60 and ADR-resistant HL-60/ADR
cell lines in vitro were compared. As shown in Fig. 2, the expression of HIF-1α, GLUT1 and
HK-II was considerably higher in HL-60/ADR cells compared with
HL-60 cells (HIF-1α, 3.70±0.084 vs. 1.00±0.075, P<0.001; GLUT1,
3.36±0.149 vs. 1.0±0.080, P<0.001; HK-II, 3.23±0.161 vs.
1.0±0.037, P<0.001), respectively.
Serum LDH level in AML patients
The level of serum LDH was 149.90±31.66,
490.63±213.94, 490.75±278.35 and 1211.57±456.99 U/l in the healthy
control, CR, PR and NR groups, respectively. The serum LDH level
was significantly increased in the NR group compared with the
control (P<0.001), CR (P<0.001) and PR groups (P=0.003).
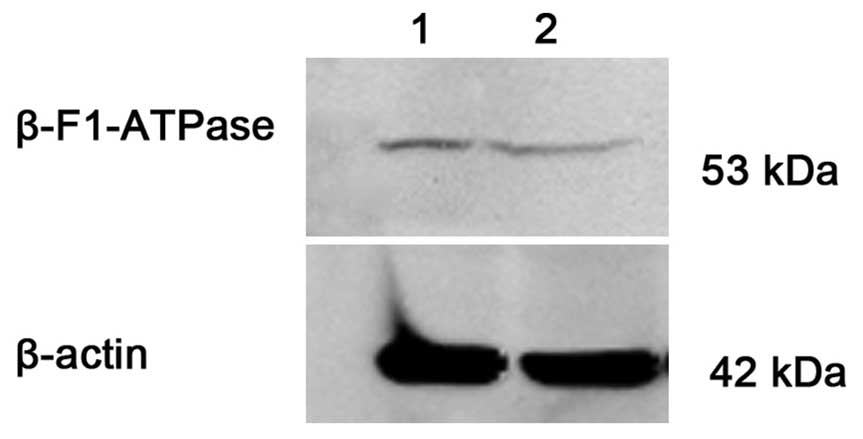
β-F1-ATPase protein expression in
primary AML cells and cell lines
The protein expression level of β-F1-ATPase/β-actin
was 0.0540±0.0482 and 0.0092±0.0042 in the CR (n=20) and NR (n=12)
groups, respectively. There was a significant difference between
the two groups (P=0.017). Consistently, ADR-resistant HL-60/ADR
cells showed a lower β-F1-ATPase expression compared with
ADR-sensitive HL-60 cells (Fig.
3).
Effect of glycolytic inhibitor on
cytotoxicity in AML cell lines
The present study investigated the response of the
AML HL-60 and HL-60/ADR cell lines to ADR while inhibiting the
glycolysis pathway. The cells were incubated with 2-DG or 3BrPA,
which resulted in a proximal blockade of glycolysis (Fig. 4A). Following 4 days of treatment of
the HL-60 and HL-60/ADR cell lines with different concentrations of
2-DG (HL-60, 0.5 mM; HL-60/ADR, 2 mM) or 3BrPA (HL-60, 20 µM;
HL-60/ADR, 20 µM), either alone or in combination with ADR (HL-60,
0.001 µg/ml; HL-60/ADR, 0.5 µg/ml), a considerable reduction of
glucose uptake was identified compared with non-treated cells
(Fig. 4A). The decrease in glucose
consumption was not observed when cells were incubated with ADR
alone.
In addition, the combination of 2-DG or 3BrPA and
ADR resulted in markedly increased cytotoxicity in ADR-resistant
HL-60/ADR cells (CDI <0.7), compared with treatment with
glycolysis inhibitor or ADR as individual drugs (Fig. 4B). However, this synergistic effect on
cell death was not observed in ADR-sensitive HL-60 cells (CDI
>0.7 and <1).
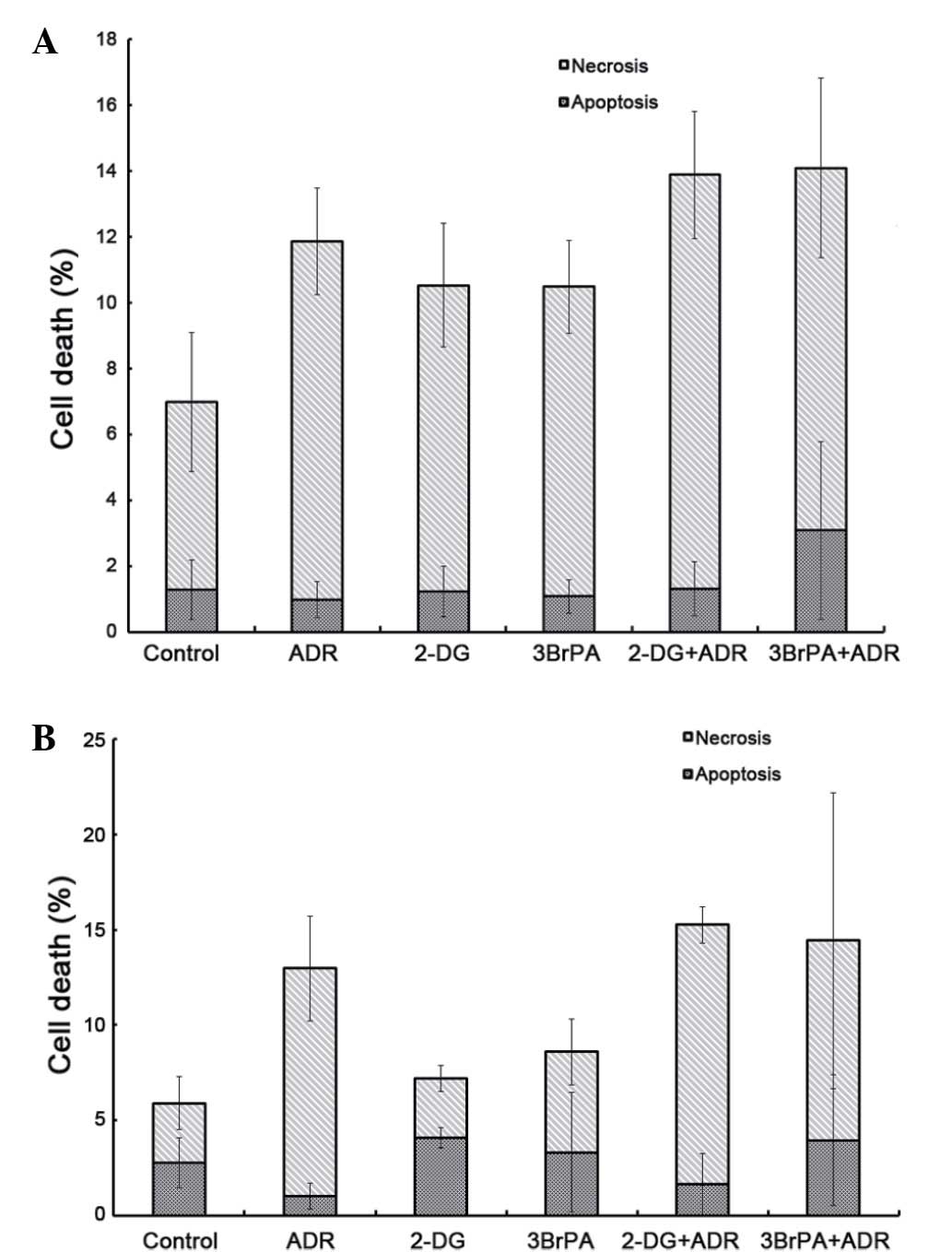
Effect of glycolytic inhibitors on
apoptosis in AML cell lines
A flow cytometry assay analyzed the apoptotic rates
of HL-60 and HL-60/ADR cells treated with glycolytic inhibitor
(2-DG or 3BrPA) or ADR or a combination of the two agents.
Following 24 h of treatment of the cell lines with 2-DG or 3BrPA,
either alone or in combination with ADR, the percentage of
apoptotic cells in the HL-60 and HL-60/ADR cell lines showed no
significant difference (Fig. 5A;
P>0.05). However, treatment with 2-DG or 3BrPA in combination
with ADR increased the necrosis of cells (Fig. 5B; 0.001 µg/ml ADM vs. control,
P=0.012; 0.5 mM 2-DG + 0.001 µg/ml ADM vs. control, P=0.003; 20 µM
3BrPA + 0.001 µg/ml ADM vs. control, P=0.017).
Discussion
Resistance to chemotherapeutic agents is a major
obstacle to the successful treatment of AML. Therefore, the
development of strategies to reverse resistance to these agents may
enhance the efficacy of AML treatment. Several potential mechanisms
for resistance to chemotherapeutic agents have been studied in
leukemia cell lines and patients with AML, but the majority of the
mechanisms were not identified as sufficient causes of resistance
(23). The extrusion of drugs by
membrane ATP-dependent drug efflux pumps, termed ATP-binding
cassette transporters, is commonly implicated in MDR. Other drug
efflux pumps include P-gp, MRP, Topo II and LRP, which also
contribute to chemoresistance in AML and have not yet been shown to
be effective in clinical trials (24,25).
Previous studies have revealed that enhanced glucose uptake and
aerobic glycolysis is one of the fundamental phenotypes of
malignant tumors, and is important for tumor relapse and
chemoresistance (26,27). It has been reported that
drug-resistant cells are characterized by defective mitochondrial
ATP production, elevated aerobic glycolysis, increased absolute
levels of intracellular ATP and enhanced HIF-1α-mediated signaling
in tumor cells (27). Similar to the
findings in solid tumors (28), it
has been reported that high glycolytic metabolism was associated
with resistance to the induction of apoptosis by chemotherapeutic
agents in AML primary cells and cell lines (11,29). The
present results showed that the elevated glucose consumption in the
ADR-resistant AML HL-60/ADR cell line compared with ADR-sensitive
HL-60 cells was consistent with previous findings (11). The percentage of apoptotic cells in
these cell lines showed no significant difference subsequent to
treatment with a glycolysis inhibitor alone or in combination with
ADR. However, 2-DG or 3BrPA in combination with ADR increased the
necrosis in these cell lines (P<0.05). Based on these initial
findings, the present study suggested that a high level of
glycolysis may be associated with chemotherapy resistance in
AML.
It has been shown that cancer cells, including
leukemia cells, shift energy production from oxidative
phosphorylation towards the less efficient glycolysis pathway
(5–11,26–28). The
increased glycolysis rate is due to the upregulation of several
genes involved in glycolysis or glucose uptake. This upregulation
in gene expression is generally considered to be due to the
activation of the transcription factors c-MYC or HIF-1α (12). Previous data suggests that
HIF-1α-mediated signaling plays a central role in the process of
glycolysis and chemoresistance in tumors (27). HIF-1α is a transcriptional factor that
upregulates the expression of specific isoforms of GLUT, HK, PFK-1,
PFK-2, aldolase, GAPDH, phosphoglycerate kinase, phosphoglycerate
mutase, ENO, pyruvate kinase and LDH (15). Consequently, the glycolytic flux and
the levels of intermediaries are increased.
Reviewing the biochemistry of glycolysis revealed
that GLUT, HK and LDH are the main controlling steps of the
glycolytic flux in cells (15,30,31).
It appears that such modifications in these and other glycolytic
steps are a component of the mechanisms involved in the increased
glycolytic flux of tumor cells. The first rate-limiting step of
glucose metabolism is the transport of glucose across the plasma
membrane. The GLUT family of proteins is responsible for this
transport, and is often found to be dysregulated or overexpressed
in malignant cells (30). HK-II
catalyzes the first step in the glycolytic pathway, which
phosphorylates glucose into glucose-6-phosphate in the glycolytic
pathway (31). LDH catalyzes the
conversion of pyruvate and NADH to lactate and NAD+,
which is the final step in the glycolytic pathway, and has a
critical role in tumor maintenance (30). Several studies have demonstrated an
association between the expression of GLUT1, HK and LDH and
chemoresistance in tumors (32–34). A
previous study has shown that hypoxia is closely associated with
tumor resistance to anticancer drugs, and numerous factors
contribute to the mechanisms of resistance (35). Direct and indirect factors of hypoxia
account for drug resistance. The present study demonstrated that
the expression of HIF-1α, GLUT1, HK-II and LDH was significantly
increased in AML patients with NR compared with patients with CR
and PR. Similarly, the expression of HIF-1α, GLUT1 and HK-II in
HL-60/ADR cells was also increased compared with the expression in
HL-60 cells. These findings suggested that HIF-1α-mediated
signaling was associated with chemoresistance in AML, and the
altered expression of a series of genes involved in glucose
metabolism may collectively contribute to glycolysis.
In certain tumors, this glycolytic phenotype is
accompanied by a loss of bioenergetic activity in mitochondria
(36–38). β-F1-ATPase is the β catalytic subunit
of mitochondrial H+-ATP synthase and is the key enzyme
in ATP synthesis (37). Previous
studies demonstrated that downregulation of β-F1-ATPase was
associated with resistance of cancer cells to anticancer therapies
(38,39). In the present study, it was found that
the level of the β-F1-ATPase protein in ADR-resistant HL-60/ADR
cells was markedly decreased compared with the level in
ADR-sensitive HL-60 cells. Similarly, β-F1-ATPase expression was
decreased in the NR group compared with the CR group. Overall,
these results indicated the importance of the damaged mitochondrial
oxidative phosphorylation regulated by β-F1-ATPase in the
chemoresistance of AML.
Since drug-resistant cancer cells have increased
glycolysis metabolism compared with drug-sensitive cancer and
normal cells, studies have hypothesized that inhibition of
glycolysis may overcome drug resistance in drug-resistant cancer
cells (6–13). A previous study found that 3BrPA
enhanced the cytotoxic effect of ADR, vincristine and Ara-C in
drug-resistant HL-60/ADR cells, which was consistent with the
aforementioned hypothesis (18).
Chemopotentiating effects were investigated in ovarian carcinoma
cells. The higher the resistance of a cell line to platinum, the
more sensitive the cell line was to potentiation of platinum using
DG, and increased glucose metabolism was shown (38). Similarly, glycolytic inhibitors, such
as 2-DG, LND and 3BrPA, may reverse glucocorticoid resistance in
prednisolone-resistant ALL cell lines (12). The present study showed that
specifically blocking the glycolytic pathway affected ADR-resistant
AML cells with high glycolytic activity, while no evident
sensitizing effect was observed in cells that were already
sensitive to ADR. This synergistic effect rendered ADR-resistant
leukemia cells more susceptible to ADR.
It has been reported that the development of
chemoresistance is associated with alterations in the apoptotic
cell death pathway (40). There is
also evidence to suggest that the anticancer effects of glycolytic
inhibitors may be due to the activation of non-apoptotic cell death
(41). Rapamycin and 2-DG markedly
enhance apoptotic and non-apoptotic cell death in ovarian tumor
cells (41). In the present study,
the administration of 3BrPA or 2-DG was also found to enhance
ADR-induced cytotoxic effects mediated by non-apoptotic cell death,
in addition to apoptosis. The underlying mechanism of cell death
may be a decrease in intracellular ATP levels induced by glycolytic
inhibitors and ADR, which consequently induces non-apoptotic cell
death. By reducing intracellular ATP levels, 2-DG may prevent
ovarian cancer cells from using ATP-dependent transporters,
including P-gp, which are a major cause of MDR. The notable
decrease in intracellular ATP levels may render cells unable to
remove cytotoxic drugs from the cell, resulting in increased
intracellular concentrations of the drug and increased cell death.
In the present study, treatment with 3BrPA or 2-DG in combination
with ADR resulted in reduced glucose consumption. This may cause
insufficient synthesis of ATP, which also supports the present
hypothesis.
In conclusion, we the present study showed that drug
resistance in AML cells was associated with increased glycolytic
activity and low efficiency of oxidative phosphorylation, which was
observed by the elevated expression of glycolysis-associated
molecules such as HIF-1α, HK-II, GLUT1 and LDH. Inhibition of
glycolysis may be a promising approach to reverse chemoresistance
in patients with AML.
Acknowledgements
The present study was supported by grants from the
Postdoctoral Science Foundation of China (grant no. 20100480834),
Natural Science Foundation of Guangdong Province, China (grant no.
S2012010008865) and Medical Science Foundation of Guangdong
Province, China (grant no. B2010340).
References
|
1
|
Estey EH: Acute myeloid leukemia: 2013
update on risk-stratification and management. Am J Hematol.
88:318–327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shipley JL and Butera JN: Acute
myelogenous leukemia. Hematol. 37:649–658. 2009.
|
|
3
|
Shaffer BC, Gillet JP, Patel C, Baer MR,
Bates SE and Gottesman MM: Drug resistance: Still a daunting
challenge to the successful treatment of AML. Drug Resist Updat.
15:62–69. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Andreeff M and Konopleva M: Mechanisms of
drug resistance in AML. Cancer Treat Res. 112:237–262. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Holleman A, Cheok MH, den Boer ML, Yang W,
Veerman AJ, Kazemier KM, Pei D, Cheng C, Pui CH, Relling MV, et al:
Gene-expression patterns in drug-resistant acute lymphoblastic
leukemia cells and response to treatment. N Engl J Med.
351:533–542. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beesley AH, Firth MJ, Ford J, Weller RE,
Freitas JR, Perera KU and Kees UR: Glucocorticoid resistance in
T-lineage acute lymphoblastic leukaemia is associated with a
proliferative metabolism. Br J Cancer. 100:1926–1936. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao F, Mancuso A, Bui TV, Tong X, Gruber
JJ, Swider CR, Sanchez PV, Lum JJ, Sayed N, Melo JV, et al:
Imatinib resistance associated with BCR-ABL upregulation is
dependent on HIF-1alpha-induced metabolic reprograming. Oncogene.
29:2962–2972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kluza J, Jendoubi M, Ballot C, Dammak A,
Jonneaux A, Idziorek T, Joha S, Dauphin V, Malet-Martino M,
Balayssac S, et al: Exploiting mitochondrial dysfunction for
effective elimination of imatinib-resistant leukemic cells. PLoS
One. 6:e219242011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kominsky DJ, Klawitter J, Brown JL, Boros
LG, Melo JV, Eckhardt SG and Serkova NJ: Abnormalities in glucose
uptake and metabolism in imatinib-resistant human BCR-ABL-positive
cells. Clin Cancer Res. 15:3442–3450. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klawitter J, Kominsky DJ, Brown JL,
Klawitter J, Christians U, Leibfritz D, Melo JV, Eckhardt SG and
Serkova NJ: Metabolic characteristics of imatinib resistance in
chronic myeloid leukaemia cells. Br J Pharmacol. 158:588–600. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Herst PM, Howman RA, Neeson PJ, Berridge
MV and Ritchie DS: The level of glycolytic metabolism in acute
myeloid leukemia blasts at diagnosis is prognostic for clinical
outcome. J Leukoc Biol. 89:51–55. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hulleman E, Kazemier KM, Holleman A,
VanderWeele DJ, Rudin CM, Broekhuis MJ, Evans WE, Pieters R and Den
Boer ML: Inhibition of glycolysis modulates prednisolone resistance
in acute lymphoblastic leukemia cells. Blood. 113:2014–2021. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akers LJ, Fang W, Levy AG, Franklin AR,
Huang P and Zweidler-McKay PA: Targeting glycolysis in leukemia: A
novel inhibitor 3-BrOP in combination with rapamycin. Leuk Res.
35:814–820. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pradelli LA, Bénéteau M, Chauvin C,
Jacquin MA, Marchetti S, Muñoz-Pinedo C, Auberger P, Pende M and
Ricci JE: Glycolysis inhibition sensitizes tumor cells to death
receptors-induced apoptosis by AMP kinase activation leading to
Mcl-1 block in translation. Oncogene. 29:1641–1652. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moreno-Sánchez R, Rodríguez-Enríquez S,
Saavedra E, Marín-Hernández A and Gallardo-Pérez JC: The
bioenergetics of cancer: Is glycolysis the main ATP supplier in all
tumor cells? Biofactors. 35:209–225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen JQ and Russo J: Dysregulation of
glucose transport, glycolysis, TCA cycle and glutaminolysis by
oncogenes and tumor suppressors in cancer cells. Biochim Biophys
Acta. 1826:370–384. 2012.PubMed/NCBI
|
|
17
|
Gillies RJ, Robey I and Gatenby RA: Causes
and consequences of increased glucose metabolism of cancers. J Nucl
Med. 49(Suppl 2): S24–S42. 2008. View Article : Google Scholar
|
|
18
|
Xu RH, Pelicano H, Zhou Y, Carew JS, Feng
L, Bhalla KN, Keating MJ and Huang P: Inhibition of glycolysis in
cancer cells: A novel strategy to overcome drug resistance
associated with mitochondrial respiratory defect and hypoxia.
Cancer Res. 65:613–621. 2005.PubMed/NCBI
|
|
19
|
World Medical Association: World Medical
Association Declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen CY, Jia JH, Zhang MX, Meng YS, Kong
DX, Pan XL and Yu XP: Proteomic analysis on multi-drug resistant
cells HL-60/DOX of acute myeloblastic leukemia. Chin J Physiol.
48:115–120. 2005.PubMed/NCBI
|
|
21
|
Wang D, Wang Z, Tian B, Li X, Li S and
Tian Y: Two hour exposure to sodium butyrate sensitizes bladder
cancer to anticancer drugs. Int J Urol. 15:435–441. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chinese Chemotherapy Symposium of
Leukemia: The chemotherapy response criteria of acute leukemia
Chin. J Hematol. 9:183–184. 1988.
|
|
23
|
Reagan JL, Fast LD, Safran H, Nevola M,
Winer ES, Castillo JJ, Butera JN, Quesenberry MI, Young CT and
Quesenberry PJ: Cellular immunotherapy for refractory hematological
malignancies. J Transl Med. 11:1502013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de Figueiredo-Pontes LL, Pintão MC,
Oliveira LC, Dalmazzo LF, Jácomo RH, Garcia AB, Falcão RP and Rego
EM: Determination of P-glycoprotein, MDR-related protein 1, breast
cancer resistance protein, and lung-resistance protein expression
in leukemic stem cells of acute myeloid leukemia. Cytometry B Clin
Cytom. 74:163–168. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaufmann SH, Karp JE, Jones RJ, Miller CB,
Schneider E, Zwelling LA, Cowan K, Wendel K and Burke PJ:
Topoisomerase II levels and drug sensitivity in adult acute
myelogenous leukemia. Blood. 83:517–530. 1994.PubMed/NCBI
|
|
26
|
Suh DH, Kim MK, No JH, Chung HH and Song
YS: Metabolic approaches to overcoming chemoresistance in ovarian
cancer. Ann N Y Acad Sci. 1229:53–60. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou Y, Tozzi F, Chen J, Fan F, Xia L,
Wang J, Gao G, Zhang A, Xia X, Brasher H, et al: Intracellular ATP
levels are a pivotal determinant of chemoresistance in colon cancer
cells. Cancer Res. 72:304–314. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gatenby RA and Gillies RJ: Why do cancers
have high aerobic glycolysis? Nat Rev Cancer. 4:891–899. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Herst PM, Hesketh EL, Ritchie DS and
Berridge MV: Glycolytic metabolism confers resistance to combined
all-trans retinoic acid and arsenic trioxide-induced apoptosis in
HL60rho0 cells. Leuk Res. 32:327–333. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao Y, Butler EB and Tan M: Targeting
cellular metabolism to improve cancer therapeutics. Cell Death Dis.
4:e5322013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pedersen PL, Mathupala S, Rempel A,
Geschwind JF and Ko YH: Mitochondrial bound type II hexokinase: A
key player in the growth and survival of many cancers and an ideal
prospect for therapeutic intervention. Biochim Biophys Acta.
1555:14–20. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ulanovskaya OA, Cui J, Kron SJ and Kozmin
SA: A pairwise chemical genetic screen identifies new inhibitors of
glucose transport. Chem Biol. 18:222–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wolf A, Agnihotri S, Micallef J, Mukherjee
J, Sabha N, Cairns R, Hawkins C and Guha A: Hexokinase 2 is a key
mediator of aerobic glycolysis and promotes tumor growth in human
glioblastoma multiforme. J Exp Med. 208:313–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Teng CL, Young JH, Hsu SL, Chou G, Kuo IT,
Yu CY and Hwang GY: Lactate dehydrogenase, not vascular endothelial
growth factor or basic fibroblast growth factor, positively
correlates to bone marrow vascularity in acute myeloid leukemia. J
Chin Med Assoc. 69:534–537. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shannon AM, Bouchier-Hayes DJ, Condron CM
and Toomey D: Tumour hypoxia, chemotherapeutic resistance and
hypoxia-related therapies. Cancer Treat Rev. 29:297–307. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
López-Ríos F, Sánchez-Aragó M,
García-García E, Ortega AD, Berrendero JR, Pozo-Rodríguez F,
López-Encuentra A, Ballestín C and Cuezva JM: Loss of the
mitochondrial bioenergetic capacity underlies the glucose avidity
of carcinomas. Cancer Res. 67:9013–9017. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sánchez-Aragó M and Cuezva JM: The
bioenergetic signature of isogenic colon cancer cells predicts the
cell death response to treatment with 3-bromopyruvate, iodoacetate
or 5-fluorouracil. J Transl Med. 9:192011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hernlund E, Hjerpe E, Avall-Lundqvist E
and Shoshan M: Ovarian carcinoma cells with low levels of
beta-F1-ATPase are sensitive to combined platinum and
2-deoxy-D-glucose treatment. Mol Cancer Ther. 8:1916–1923. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shin YK, Yoo BC, Chang HJ, Jeon E, Hong
SH, Jung MS, Lim SJ and Park JG: Down-regulation of mitochondrial
F1F0-ATP synthase in human colon cancer cells with induced
5-fluorouracil resistance. Cancer Res. 65:3162–3170.
2005.PubMed/NCBI
|
|
40
|
Hajra KM and Liu JR: Aoptosome dysfunction
in human cancer. Apoptosis. 9:691–704. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Loar P, Wahl H, Kshirsagar M, Gossner G,
Griffith K and Liu JR: Inhibition of glycolysis enhances
cisplatin-induced apoptosis in ovarian cancer cells. Am J Obstet
Gynecol. 202(371): e1–e8. 2010.PubMed/NCBI
|