Introduction
Metastasis is considered to be one of the most
destructive characteristics of cancer. Though the causes and
genetic bases of tumorigenesis vary, the key events required for
metastasis are similar for all types of cancer, including the
alteration of adhesion ability, the enhancement of motility and the
secretion of proteolytic enzymes to degrade the basement membrane
(1,2).
The phosphatase of regenerating liver (PRL) family
of protein tyrosine phosphatases (PTPs), including PRL-1, PRL-2,
and PRL-3, emerges as potential biomarkers and therapeutic targets
for various types of malignancy (3,4). Despite
of relatively low expression in normal tissues and untransformed
cells, high expression of PRL-3 had been found in a variety of
cancer tissues, which correlates with disease progression and
survival (5–8). Reports from certain groups highlight the
oncogenic role of PRL-3 in promoting cancer metastasis through
enhanced cell motility and invasiveness (3). Further investigations have demonstrated
that PRL-3 stimulates invasiveness by activating the Rho family of
small GTPases and matrix metalloproteinase-2 (MMP-2) (9,10). PRL-3
negatively regulates C-terminal Src kinase (Csk) and PTEN, leading
to enhanced activities of Src kinase and PI3K/AKT signaling
pathways (11,12). By upregulating the activity of signal
transducers and activators of transcription (STAT) pathway and the
expression of anti-apoptotic factor Mcl-1, PRL-3 confers
therapeutic resistance to small molecule inhibitors. In addition,
as a downstream target of the tumor suppressor p53, PRL-3
negatively regulates p53 and PRL-3 modulates cell-cycle progression
through the PI3K-AKT pathway (13).
Despite of these functions, the role of PRL-3 in other key steps of
tumorigenesis in uncertain.
JAM2 (or JAM-B) belongs to the junctional adhesion
molecule (JAMs) family, which is composed of 6 immunoglobulin-like
members: CAR, ESAM, JAM4, JAM-A, JAM-B and JAM-C (14,15). The
majority of research into JAMs focuses on the relationship between
differential expression of JAMs and leukocyte movement and
redistribution. JAM-B and its family members have been associated
with endothelial cell-cell adhesion and leukocyte transmigration
through homo/heterophillic interaction. JAM-B stabilizes and
recruits JAM-C in the junction complex on the cell-cell contacts
through heterophillic interaction (16–18). Two
independent groups demonstrated that the JAM-B gene is expressed in
three stem cell lines using a DNA microarray method (18,19). The
relevance of JAMs within cancer development has rarely been
reported (20).
In the present study, the effect of PRL-3 on
adhesion and motility in the human embryonic kidney cell line 293
and the colon cancer cell line LoVo are systemically analyzed. The
molecular role of PRL-3 in cell movement and rearrangement of cell
skeleton were investigated as were the effects of overexpression of
PRL-3 on cell-matrix cell spread speed and cell-matrix adhesion. To
explore the potential mechanism of PRL-3 in cell adhesion and
movement, JAM2 was investigated as a new interaction protein of
PRL-3. The synergism of PRL-3 and JAM2 promotes cancer
cell-endothelial cell adhesion. These results provided an
indication that the function of PRL-3 in tumor metastasis may be
associated with the junctional adhesion molecules. Blocking the
interaction of PRL-3 and JAM2 maybe a new approach to inhibiting
metastasis in patients in the future.
Materials and methods
Cell lines, plasmid and antibody
Flp-In-293 (293) cell line (Invitrogen; Thermo
Fisher Scientific, Inc., Carlsbad, CA, USA) and the colon cancer
cell line LoVo (American Type Culture Collection, Manassas, VA,
USA) were cultured in Dulbecco's modified Eagles medium (DMEM) and
Ham's F12 K medium supplemented with 10% fetal bovine serum (FBS,
ThermoFisher Scientific, Inc.), respectively. LoVo cells stably
expressing PRL-3 and control cells were previously established
(10).
The eukaryon plasmid pDsRED-JAM2 (Clontech
Laboratories, Inc., Mountainview, CA, USA) was constructed in our
laboratory by inserting full length JAM2 cDNA into a vector.
pEBG-JAM2 and pCDNA-Myc-JAM2 were constructed and saved by our
laboratory previously. Monoclonal antibody (3B6) against PRL-3 was
prepared as previously reported (21).
In vitro wound healing assay
Cells were seeded onto 6-well plates at a
sub-confluent density (5×105/well). After 12 h, a line
was scraped out of the cell monolayer using a 200-µl pipet tip and
the width of this wound line was captured using an inverted
microscope (ECLIPSE TS100, Nikon, Tokyo, Japan) at a 24 h interval.
The speed of motility of the cells was assessed using the degree of
healing of the wound line. The experiment was repeated 3 times
independently.
Cell spread assay
Six-well plates were coated with 5 µg/well collagen
I (Cohesion Technologies Inc., Palo Alto, CA, USA), 1 µg/well
fibronectin (Sigma-Aldrich Corporation, St. Louis, MO, USA)
overnight at 4°C or left untreated. Next, the plates were blocked
with 2% bovine serum albumin (BSA; Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) and washed with
phosphate-buffered saline (PBS). Cells were seeded at a density of
5×104 cells per well in 6 wells and incubated for 15 min
at 37°C, then the cell morphology was observed under a light
microscope (XDS-300C; Caikon Optical Instrument Co., Ltd.,
Shanghai, China), the attached cells were counted and the
percentage of attached cells was estimated.
Cell-matrix adhesion assay
24-well plates were coated with 5 µg/well collagen I
(Cohesion Technologies Inc.), 1 µg/well fibronectin (Sigma-Aldrich)
overnight at 4°C or left untreated. Next, the plates were blocked
with 2% BSA and washed with PBS. Cells were seeded at the density
of 1×104 cells per well in 6 wells and incubated for
1,2,3 or 5 min at 37°C. Then the un-attached cells of 3 parallel
wells were discarded by gently washing 3 times with PBS. The number
of cells that unattached from the wells were evaluated by cytometry
(Cellometer Auto T4; Nexcelom Bioscience LLC, Lawrence, MA, USA)
and the adhesion rate was expressed as the percentage of the mean
amount of washed wells to that of un-washed wells.
Reverse transcription (RT)-polymerase
chain reaction (PCR)
Ec03 and HmEC cells were cultured and RNA was
extracted from cells using Invitrogen Trizol reagent (Thermo Fisher
Scientific, Inc.). RT was conducted using a Reverse Transcription
System (#A3500; Promega Corporation, Madison, WI, USA) according to
the manufacturer's instructions with the following quantities of
reagents: Total RNA, 1 µg; random primers (0.5 µg/µl), 1 µl; oligdT
(2 µg/µl), 1 µl; dNTPs (10 mM), 1 µl; 5X buffer, 4 µl; RNase
inhibitor (40 U/ml), 0.5 µl; M-MLV (200 U/ml), 1 µl;
MgCl2 (25 mM), 1 µl; and ddH2O to a total
volume of 20 µl. For PCR, the reaction mixture consisted of 1 µl
DNA, 1 µl upstream primer, 1 µl downstream primer, 12.5 µl 2X PCR
Master Mix (Thermo Fisher Scientific, Inc.), and ddH2O
to a total volume of 25 µl. Primers were purchased from Sangon
Biotech Co., Ltd. (Shanghai, China): JAM2 sense,
5′-AGCAGTAGAGTACCAAGGTGA-3′; JAM2 antisense,
5′-TACGGCTGCTATGATGCCAC-3′; GAPDH sense,
5′-CGGAGTCAACGGATTTGGTCGTAT-3′; and GAPDH antisense,
5′-AGCCTTCTCCATGGTGGTGAAGAC-3′. PCR was performed in an Applied
Biosystems 2720 Thermal Cycler (Thermo Fisher Scientific, Inc.)
using the following reaction conditions: 95°C for 5 min; 29 cycles
of 94°C for 30 sec, 58°C for 45 sec and 72°C for 40 sec; and a
final step of 72°C for 10 min. Products were stored at 4°C. PCR
products were electrophoretically separated on 0.8% agarose gels
and were visualized using GeneGenius Bio Imaging system (Syngene
Bioimaging Private Ltd., Gurgaon, India).
Western blot assay and
immunoprecipitation
Cells were seeded (1.5×105/well) and
transfected with pEBG-JAM2, pCDNA-Myc-JAM2 (4 µg) and the
respective vector for 72 h using Invitrogen Lipofectamine reagent
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions; they were then lyzed in lysis buffer (50 mM Tris-HCl,
pH 7.5, 150 mM NaCl, 1% NP-40, 1 mmol/l phenylmethylsulfonyl
fluoride, 1 µg/ml aprotinin, and 1 µg/ml pepstatin) for 20 min at
4°C. The supernatant was collected after centrifugation at 12,000 ×
g and subjected to western blotting or immunoprecipitation.
For immunoprecipitation, the supernatant was
incubated with a mouse monoclonal anti-Myc antibody (1 µg/ml;
#TA100010; OriGene Technologies, Inc., Rockville, MD, USA).
Pre-immune serum was used as control. The precipitates were washed
four times with lysis buffer and once with PBS, and eluted in 2X
loading buffer. Protein samples were resolved by sodium dodecyl
sulfate polyacrylamide gel electrophoresis and electroblotted onto
nitrocellulose membranes (Hybond-C; #RPN303C; GE Healthcare Life
Sciences, Little Chalfont, UK), which were then blocked in 5% skim
milk in PBS-Tween, and probed with the indicated antibodies [mouse
monoclonal anti-myc (#TA100010) or anti-GST (#TA150102) antibodies
(OriGene Technologies, Inc.); final concentration 1 µg/ml) at 4°C
overnight, washed with 0.1% Tween-PBS three times, then incubated
with horseradish peroxidase-linked horse anti-mouse IgG antibody
(#7076; Cell Signaling Technology, Inc., Danvers, MA, USA;
dilution, 1:2,000) at room temperature for 45 min). Protein bands
were visualized using a Pierce enhanced chemiluminescence detection
system (Thermo Fisher Scientific, Inc.).
Immunofluorescence
LoVo cells were transiently transfected wth
pDsRED-JAM2 and pEGFP-PRL-3 plasmid for 48 h, followed by 4%
paraformaldehyde fixation and counterstained with DAPI (1 µg/ml
(#ZLI-9557; Origene Technologies, Inc.). To label actin filaments,
cells were fixed with 4% paraformaldehyde and stained with 5 µg/ml
rhodamine-conjugated phalloidin (Sigma-Aldrich) in the dark for 20
min. Images were captured using a confocal microscope (Lecia TCS
SP5; Leica Microsystems GmbH, Wetzlar, Germany).
Cell-cell adhesion assay
Endothelial cells EC03 and HmEC (China
Infrastructure of Cell Line Resources, Beijing, China) were grown
on the 24-well plate (4×105/well) for 24 h, and PBS
washed 3 times with gentle shaking, then seeded LoVo cancer cells
expressing ectopic PRL-3 and control cell for indicated time point.
The cells were carefully washed and non-adhering cancer cells were
collected, and counted by hemocytometer. A total of 3 independent
experiments were repeated.
Results
PRL3 promotes colon cancer cell
motility
To examine the motility-promoting potential of
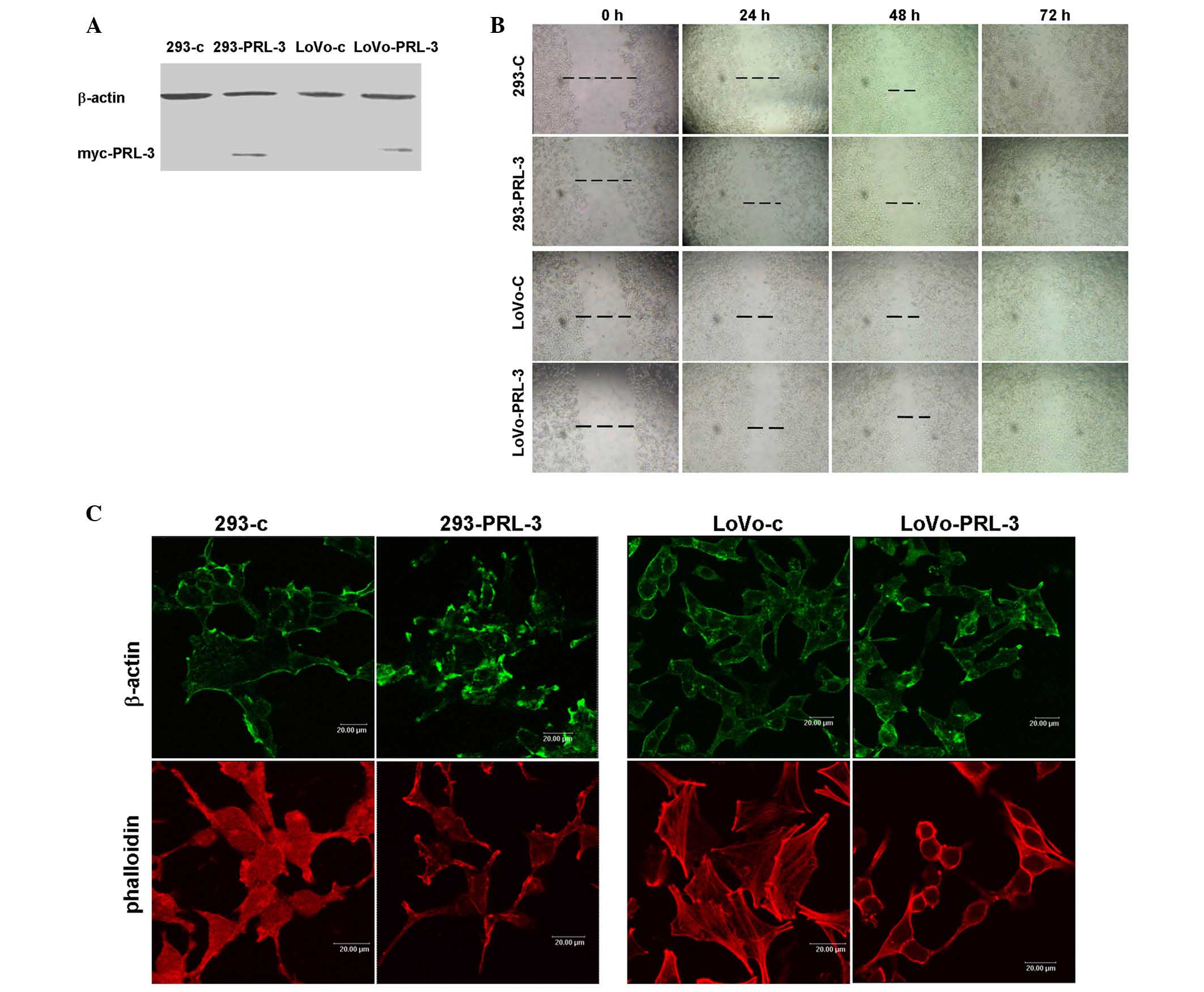
PRL-3, myc-tagged PRL-3 was stably expressed in 293 and LoVo cells
(Fig. 1A). Next, a wounding closure
assay was performed. A line was scraped through the cell monolayer
and the closure of these lines was recorded at 24 h intervals. The
results demonstrated that the speed of wound healing of 293-PRL-3
and LoVo-PRL-3 were faster than their respective control cells. A
total of 48 h or 72 h after wounding, the PRL-3 transfected cells
had moved to close the wound, while those of their control cells
remained apart (Fig. 1B).
The dynamic regulation of the actin network is
crucial for cell motility (22,23). PRL-3
has been reported to regulate the activity of the small GTPase
family Rho (11). Rho family members
serve an important role in regulating the arrangement of the actin
skeleton and pseudopodia. Therefore, the present study examined
whether the effect of PRL-3 on motility is related to its role in
actin filament remodeling. The distribution of β-actin by
immunofluorescence assay and found that β-actin was more strongly
labeled on the cell protrusions of 293-PRL-3 and LoVo-PRL-3 cells
compared to their respective control cells (Fig. 1C), indicating that PRL-3 may
participate in the rearrangement of the actin skeleton. The actin
filament distribution was stained with rhodamine
conjugated-phalloidin, a small molecular toxin that specifically
binds to filamentous actin (F-actin), but not monomeric actin. It
was observed that F-actin was enriched at the cell membrane,
particularly in the protrusion and pseudopodia in 293-PRL-3, while
diffusely distributed in 293 control cells. In LoVo cells, F-actin
was more strongly labeled in LoVo-PRL-3 cells on the protrusions of
the cell membrane compared to distribution of F-actin in LoVo
control cells. These data indicated that PRL-3 overexpression may
have induced filamentous actin remodeling to promote cell
motility.
PRL3 suppresses colon cancer cell
spread speed and cell-matrix adhesion
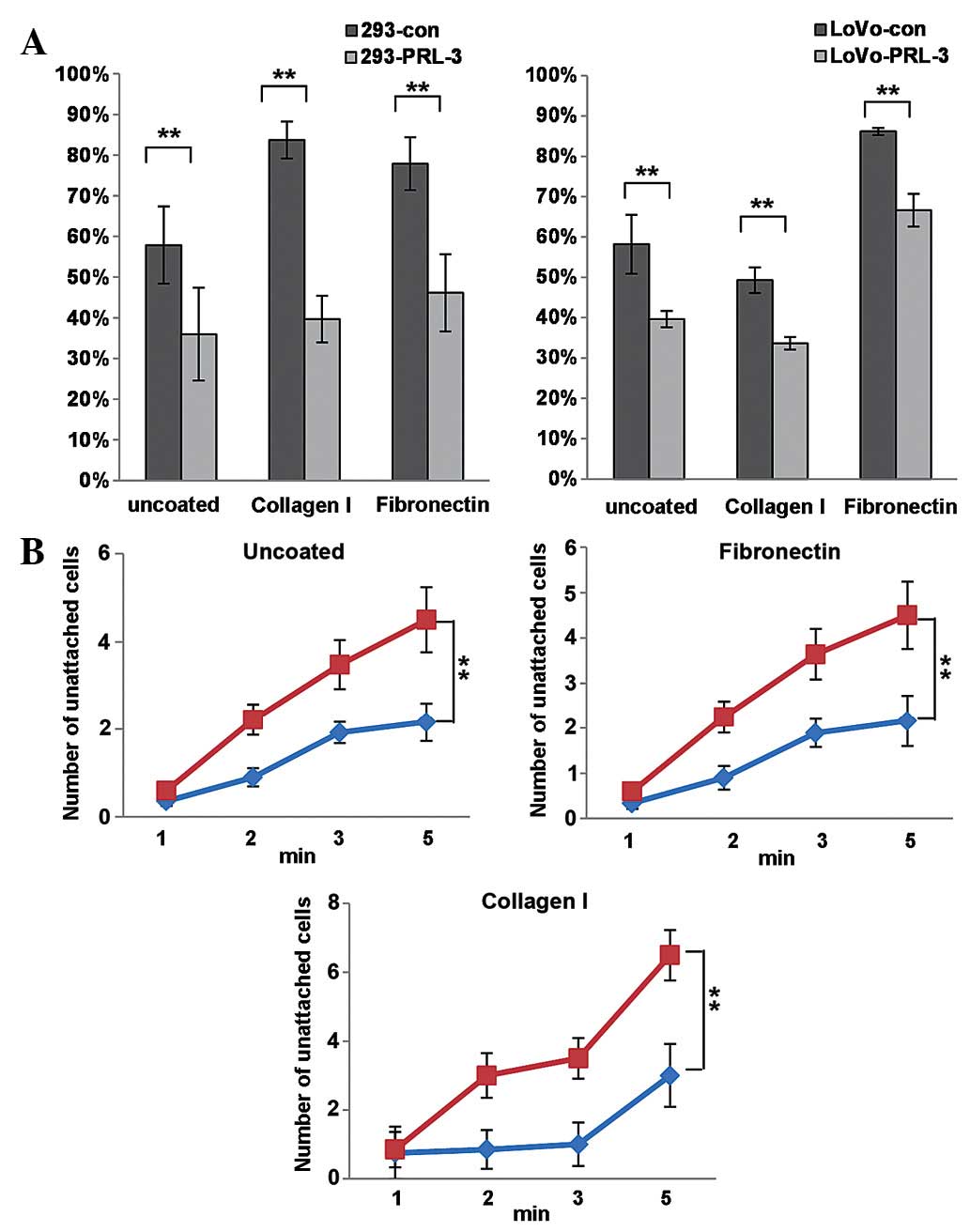
Notably, it was observed PRL-3 reduced the spread
speed of colon cancer cells (Fig.
2A). The spreading speed of control and PRL-3 transfected 293
and LoVo cells on extracellular matrix (ECM) components collagen I
and fibronectin were examined 15 min after the cells were seeded.
Spreading cells appeared as flattened and less refractive, whereas
un-spread cells were round and brighter; the percentage of
spreading cells to total cells was estimated. As presented in
Fig. 2A, 293-PRL-3 and LoVo-PRL-3
cells spread much less than their respective control cells did on
un-coated, collagen I-coated or fibronectin-coated plates
(P<0.05). Consistently, PRL-3 expression decreased the
cell-matrix adhesion in 293 and LoVo cells at the beginning time
point of EDTA-digestion. The unattached cells were counted at the
indicated time point following EDTA-treatment, the number of
unattached cells of PRL-3 overexpressing group was dramatically
higher compared to the control groups. It was concluded that PRL-3
expression promotes cell motility and actin remodeling, and PRL-3
reduces the cell spread and cell-matrix adhesion of cancer
cells.
PRL3 interacts with JAM2
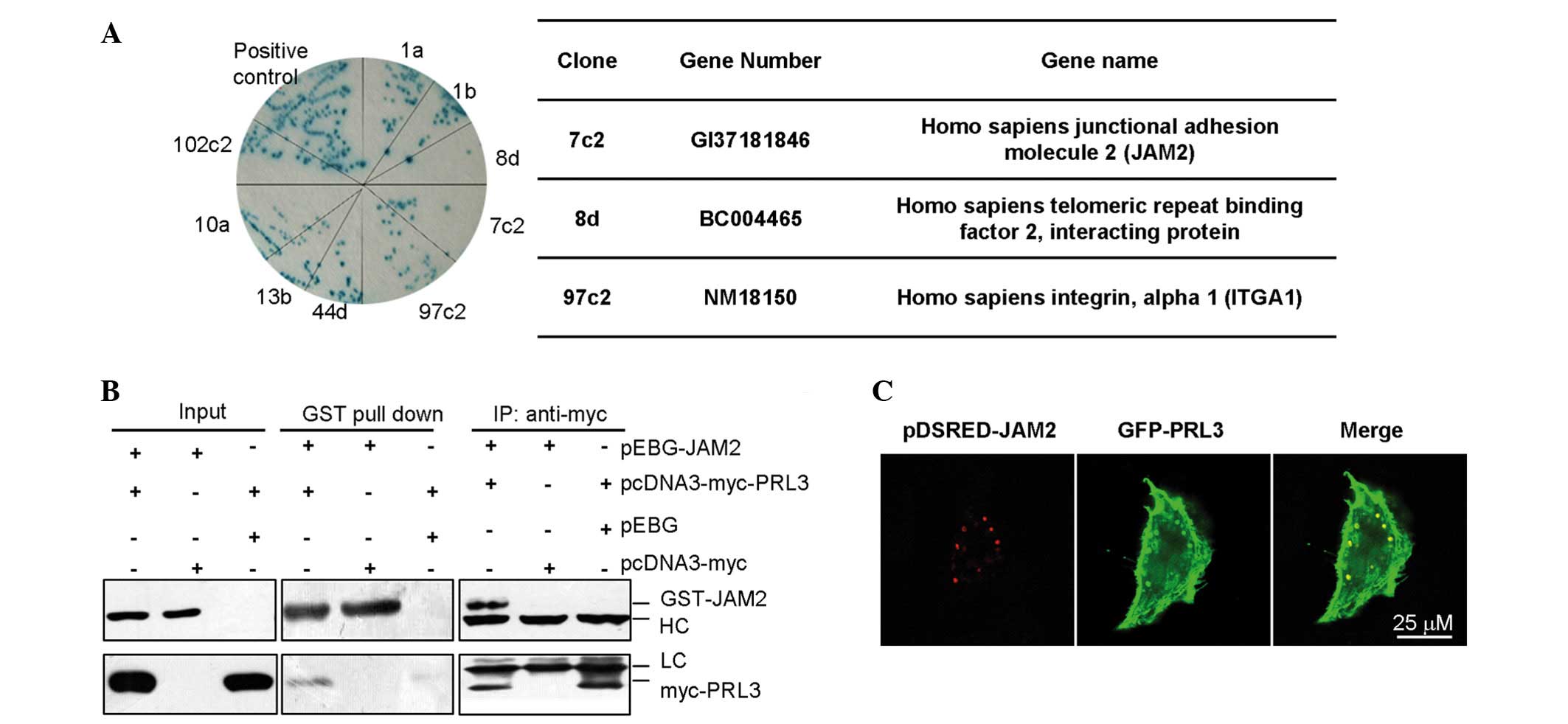
To explore the mechanism of PRL-3 in cell adhesion
and cell movement, two yeast hybrid systems were used to screen the
potential interacting protein(s) of PRL-3 (Fig. 3A). Using BD-PRL-3 fusion protein as a
bait protein to screen the embryo brain cDNA librabry, it was
demonstrated that JAM2 was a candidate interacting proteins of
PRL-3. To confirm the results of the two-yeast hybrid, the
interaction between PRL-3 and JAM2 were examined by
immunoprecipation with myc-PRL3, followed by western blot analysis
with an anti-GST antibody against GST-JAM2,. In addition, GST-JAM2
was pulled down and the precipitate was subjected to western blot
analysis using an anti-myc antibody against myc-PRL-3 (Fig. 3B). JAM2 is a known protein located on
cell membrane, and PRL-3 also locates on cell membrane and plasma.
Plasmids encoding pDsred-JAM2 and pEGFP-GFP-PLR-3 were
co-transfected into LoVo cells. After 48 h, the co-localization of
exogenous JAM2 and PRL-3 were observed in the cell membrane
(Fig. 3C), therefore, PRL-3 may be
associated with JAM2 in vitro.
PRL-3 promotes cancer cell-endothelial
cell adhesion by associating with JAM2
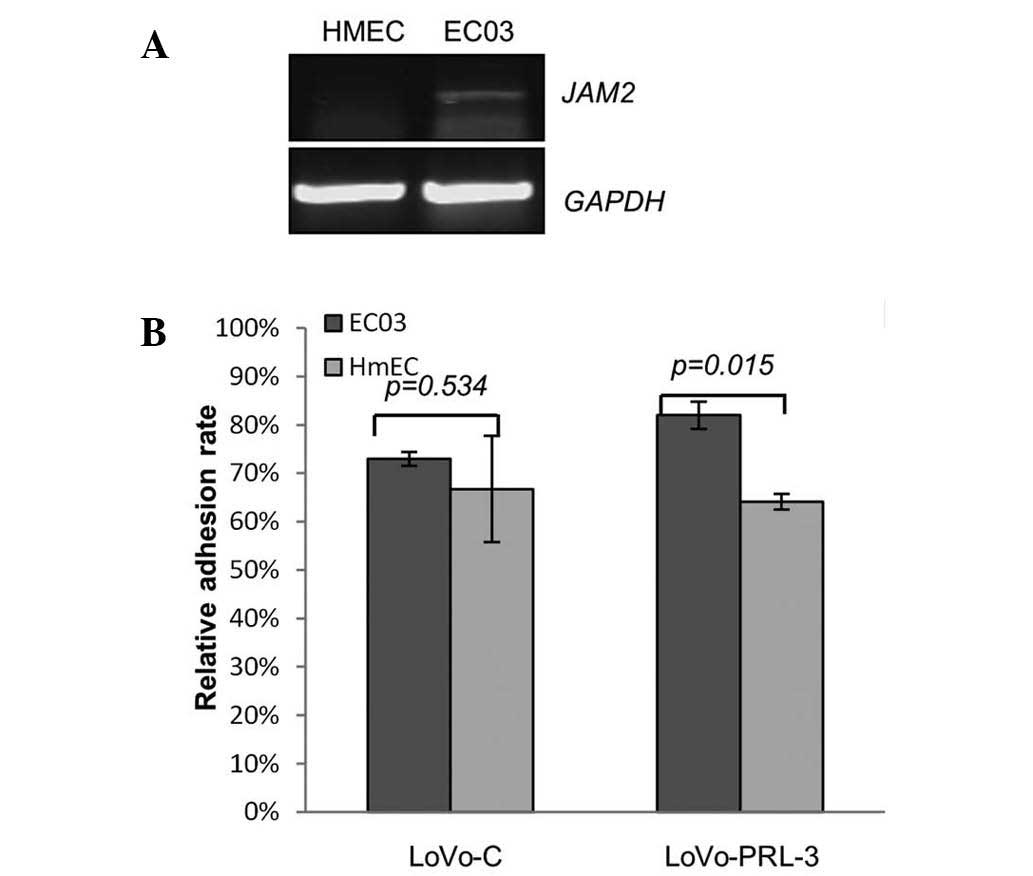
Cancer metastasis is usually a process in which
cancer cells migrate and penetrate the vascular vessels. To
investigate this process, different endothelial cell (EC03 and
HmEC) were seeded on 96 well plates, and LoVo cells expressing
ectopic PRL-3 and control cells were seeded on top and incubated
for 15 min. Following this incubation, the cells were washed with
PBS 3 times, the number of adhesive cancer cells was estimated by a
cytometer. As demonstrated in Fig. 4,
the LoVo cells expressing ectopic PRL-3 adhered much more to ECO3
cells compared with the other pairing groups. And reverse
transcription-polymerase chain reaction demonstrated that the mRNA
expression levels of JAM2 was relatively higher in EC03 cells than
in HmEC. These results indicate that ectopic PRL-3 and JAM2 may
cooperate to promote cancer cell-endothelial cell adhesion.
Discussion
Tumor metastasis is a dynamic process involving
proliferation of a primary tumor, protrusion of primary lesion, and
anchoring onto a secondary site. Cancer cell transport and
anchoring on the secondary sites through increased cell-matrix
adhesion is a key step for the metastasis (1,3,26). To survive and grow on the secondary
sites, cancer cells exhibit more pseudopodia on the cell surface
and the cytoskeleton adapts to fasten on the secondary sites
(1).
The molecular function of PRL-3 involves its
participation in the cancer metastasis process by increasing cancer
cell migration and invasion (3). The
PI3K-AKT signal pathway is also involved in the process of cell
migration and invasion induced by PRL-3. PRL-3 has also been shown
to activate EGFR, Src, ERK, JNK, and PI3K-AKT signaling (3,24,25). The role of PRL-3 in the cell movement
and cell adhesion is unclear and has not been demonstrated at
present.
In the present study, the influence of PRL-3 on the
cell motility was observed using a cell wounding healing assay and
cell spread assay (Figs. 1B and
2A). One may infer from the results
that the stable expression of PRL-3 promotes cancer cell-cancer
cell adhesion. Our results showed that PRL-3 promotes cell-cell
adhesion and gathering of cancer cells. Although the effect of
PRL-3 on the proliferation rate of cancer cells is unclear
(3,27,28), the
assembled cancer cells with relatively higher expression of PRL-3
have a stronger capability of migration, invasion and autophagy
(27). The results indicated that
expression of PRL-3 in colon cancer cells aids the survival of
primary tumor cells. However, the survival and settlement in the
distal organs is the second step in the process of tumor
metastasis. Expression of PRL-3 in the colon cancer cells
redistributed the cell skeleton protein, forming additional
pseudopodia around the cell membrane. The cell matrix binding
ability of PRL-3 expressing cells on the uncoated, fibronectin or
collagen coated plates were all markedly reduced compared with the
control cells. The cancer cells with PRL-3 expression were more
easily detached from the coated plates. In conclusion, PRL-3 may
serve an important function in the process of primary tumor
formation and protrusion. To explore the mechanism of PRL-3 in the
cell adhesion process, novel interacting proteins of PRL-3 were
identified using a yeast hybrid system. Immunopreciptation and GST
pull down assay confirmed the interaction between PRL-3 and
JAM2.
JAM2 is a protein that associates with tight
junctions and enhances homing of lymphocytes to the secondary lymph
nodes (15). JAM2 is also an
important molecule in the regulation of immune responses and
leukocyte migration. JAM2 is localized to the cell-cell tight
junction and serves a role in the maintenance of endothelial cell
architecture (13). As mentioned
above, PRL-3 reduces the spread speed and promotes the motility of
colon cancer cells (Fig. 1), the
present study hypothesiszed that PRL3 expression promotes cancer
cells to migrate to secondary sites by increasing cell motility;
after homing, PRL3 may promote cancer cell adhesion and invasion on
the endothelial cells by associating with JAM2. PRL-3-JAM2 forms
the co-localized focal in the cell endomembrane (Fig. 3C). The co-localized focal may aid the
PRL-3 expressing cancer cells to anchor and penetrate the vascular
endothelial cells. The endothelial cell-cancer cell adhesion assay
indicated that PRL-3 expression may increase cell-cell adhesion in
the presence of JAM2 expression. Then, the protrusion of primary
lesions requires the synergistic action of PRL-3 and JAM2.
Besides the well known function of PRL-3 in the
migration and invasion process of colon cancer, the newly
identified functions of PRL-3 involve the process of tumor
metastasis, particularly the process of cell matrix penetration by
tumor cells. Interrupting the interaction between PRL-3 and JAM2
may block the adhesion of vascular endothelial cells and cancer
cells. Then the cancer cells may be limited to proliferation in the
primary lesions, and the distal metastasis would be reduced.
Therefore, the disrupting the interaction between PRL-3 and JAM2
may become a potential target to prevent colon cancer
metastasis.
Acknowledgements
This work was supported by the National Natural
Science Foundation of China (grant no. 81301747).
References
|
1
|
Wan L, Pantel K and Kang Y: Tumor
metastasis: Moving new biological insights into the clinic. Nat
Med. 19:1450–1464. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maryáš J, Faktor J, Dvořáková M,
Struhárová I, Grell P and Bouchal P: Proteomics in investigation of
cancer metastasis: Functional and clinical consequences and
methodological challenges. Proteomics. 14:426–440. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al-Aidaroos AQ and Zeng Q: PRL-3
phosphatase and cancer metastasis. J Cell Biochem. 111:1087–1098.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bessette DC, Qiu D and Pallen CJ: PRL
PTPs: Mediators and markers of cancer progression. Cancer
Metastasis Rev. 27:231–252. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saha S, Bardelli A, Buckhaults P,
Velculescu VE, Rago C, St Croix B, Romans KE, Choti MA, Lengauer C,
Kinzler KW and Vogelstein B: A phosphatase associated with
metastasis of colorectal cancer. Science. 294:1343–1346. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xing X, Peng L, Qu L, Ren T, Dong B, Su X
and Shou C: Prognostic value of PRL-3 overexpression in early
stages of colonic cancer. Histopathology. 54:309–318. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peng L, Ning J, Meng L and Shou C: The
association of the expression level of protein tyrosine phosphatase
PRL-3 protein with liver metastasis and prognosis of patients with
colorectal cancer. J Cancer Res Clin Oncol. 130:521–526. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Z, Zhan W, Wang Z, Zhu B, He Y, Peng J,
Cai S and Ma J: Inhibition of PRL-3 gene expression in gastric
cancer cell line SGC7901 via microRNA suppressed reduces peritoneal
metastasis. Biochem Biophys Res Commun. 348:229–237. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fiordalisi JJ, Keller PJ and Cox AD: PRL
tyrosine phosphatases regulate rho family GTPases to promote
invasion and motility. Cancer Res. 66:3153–3161. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peng L, Xing X, Li W, Qu L, Meng L, Lian
S, Jiang B, Wu J and Shou C: PRL-3 promotes the motility, invasion,
and metastasis of LoVo colon cancer cells through PRL-3-integrin
beta1-ERK1/2 and-MMP2 signaling. Mol Cancer. 8:1102009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liang F, Luo Y, Dong Y, Walls CD, Liang J,
Jiang HY, Sanford JR, Wek RC and Zhang ZY: Translational control of
C-terminal Src kinase (Csk) expression by PRL3 phosphatase. J Biol
Chem. 283:10339–10346. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang H, Quah SY, Dong JM, Manser E, Tang
JP and Zeng Q: PRL3 down regulates PTEN expression and signals
through PI3K promote epithelial-mesenchymal transition. Cancer Res.
67:2922–2926. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bazzoni G: The JAM family of junctional
adhesion molecules. Curr Opin Cell Biol. 15:525–530. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bradfield PF, Scheiermann C, Nourshargh S,
Ody C, Luscinskas FW, Rainger GE, Nash GB, Miljkovic-Licina M,
Aurrand-Lions M and Imhof BA: JAM-C regulates unidirectional
monocyte transendothelial migration in inflammation. Blood.
110:2545–2555. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arcangeli ML, Frontera V, Bardin F,
Obrados E, Adams S, Chabannon C, Schiff C, Mancini SJ, Adams RH and
Aurrand-Lions M: JAM-B regulates maintenance of hematopoietic stem
cells in the bone marrow. Blood. 118:4609–4619. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Doñate C, Ody C, McKee T, Ruault-Jungblut
S, Fischer N, Ropraz P, Imhof BA and Matthes T: Homing of human B
cells to lymphoid organs and B-cell lymphoma engraftment are
controlled by cell adhesion molecule JAM-C. Cancer Res. 73:640–651.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lamagna C, Meda P, Mandicourt G, Brown J,
Gilbert RJ, Jones EY, Kiefer F, Ruga P, Imhof BA and Aurrand-Lions
M: Dual interaction of JAM-C with JAM-B and alpha(M)beta2 integrin:
Function in junctional complexes and leukocyte adhesion. Mol Biol
Cell. 16:4992–5003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sakaguchi T, Nishimoto M, Miyagi S, Iwama
A, Morita Y, Iwamori N, Nakauchi H, Kiyonari H, Muramatsu M and
Okuda A: Putative ‘stemnessʼ gene jam-B is not required for
maintenance of stem cell state in embryonic, neural, or
hematopoietic stem cells. Mol Cell Biol. 26:6557–6570. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arcangeli ML, Bardin F, Frontera V, Bidaut
G, Obrados E, Adams RH, Chabannon C and Aurrand-Lions M: Function
of Jam-B/Jam-C interaction in homing and mobilization of human and
mouse hematopoietic stem and progenitor cells. Stem Cells.
32:1043–1054. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morgan C, Jenkins SA, Kynaston HG and Doak
SH: The role of adhesion molecules as biomarkers for the aggressive
prostate cancer phenotype. PLoS One. 8:e816662013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peng L, Li Y, Meng L and Shou C:
Preparation and characterization of monoclonal antibody against
protein tyrosine phosphatase PRL-3. Hybrid Hybridomics. 23:23–27.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fife CM, McCarroll JA and Kavallaris M:
Movers and shakers: cell cytoskeleton in cancer metastasis. Br J
Pharmacol. 171:5507–5523. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Basak S, Jacobs SB, Krieg AJ, Pathak N,
Zeng Q, Kaldis P, Giaccia AJ and Attardi LD: The
metastasis-associated gene Prl-3 is a p53 target involved in
cell-cycle regulation. Mol Cell. 30:303–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rios P, Li X and Köhn M: Molecular
mechanisms of the PRL phosphatases. FEBS J. 280:505–524. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Al-Aidaroos AQ, Yuen HF, Guo K, Zhang SD,
Chung TH, Chng WJ and Zeng Q: Metastasis-associated PRL-3 induces
EGFR activation and addiction in cancer cells. J Clin Invest.
123:3459–3471. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
11:1423–1437. 2013. View
Article : Google Scholar
|
|
27
|
Huang YH, Al-Aidaroos AQ, Yuen HF, Zhang
SD, Shen HM, Rozycka E, McCrudden CM, Tergaonkar V, Gupta A, Lin
YB, et al: A role of autophagy in PTP4A3-driven cancer progression.
Autophagy. 10:1787–1800. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang J, Xiao Z, Lai D, Sun J, He C, Chu
Z, Ye H, Chen S and Wang J: miR-21, miR-17 and miR-19a induced by
phosphatase of regenerating liver-3 promote the proliferation and
metastasis of colon cancer. Br J Cancer. 107:352–359. 2012.
View Article : Google Scholar : PubMed/NCBI
|