Introduction
Angiogenesis is an important physiological process,
through which novel blood vessels develop from existing vessels
(1). Persistent and uncontrolled
angiogenesis is involved in the pathogenesis of rheumatoid
arthritis, atherosclerosis, diabetes, ocular retinopathy and tumors
(2,3).
In particular, tumor angiogenesis is crucial for solid tumor
growth, invasion and metastasis (4).
During tumor growth, the production of angiogenic factors,
including members of the vascular endothelial growth factor (VEGF)
and fibroblast growth factor (FGF) families, from tumor cells
results in the induction of capillary spouting and the subsequent
growth of novel vessels in tumors from surrounding host vessels
(3,5).
As the vessels in the tumor are tortuous, with sluggish flow and a
lack of surrounding pericytes, tumor cells readily invade into
novel vessels and form tumor emboli (3). In addition, tumor cells seed in distance
organs, where they undergo secondary angiogenesis. Therefore, the
disruption of angiogenesis has been considered as an effective
therapeutic strategy in the treatment of solid tumors (6).
Angiogenesis is a multi-step process involving
endothelial cell activation, proliferation, migration,
differentiation, maturation and tube formation (3,7).
Endothelial cells are central for angiogenesis. Previous studies
have demonstrated that the inhibition of endothelial cell death is
an essential prerequisite to maintain vascular remodeling and
angiogenesis (8). There are several
types of morphological distinct cell death exhibited by endothelial
cells (9). Adhesion to the
extracellular matrix (ECM) is a crucial for survival of endothelial
cells (7). During tumor angiogenesis,
the vascular basement membrane is degraded after endothelial cell
activation, and then the endothelial cells migrate into the
subendothelial space without attachment to the ECM (8). Similarly to other anchorage-dependent
cells, endothelial cells undergo cell death after detachment from
the underlying ECM, which is defined as anoikis (10).
Anti-malarial agent dihydroartemisinin (DHA) is a
semi-synthetic derivative of artemisinin that is extracted from the
herbaceous plant, Artemisia annua (11). DHA exhibits potent antitumor and
anti-angiogenesis effects and has therefore emerged as a potential
component for cancer chemotherapies (12). DHA inhibits endothelial cell
proliferation and migration via the downregulation of the nuclear
factor-κB and extracellular signal-regulated kinase (ERK) signaling
pathways (13–15). Several studies have suggested that the
anti-angiogenic effects of DHA may be partly associated with its
role in promoting the apoptosis of endothelial cells (12,16).
However, the effects of DHA on endothelial cell anoikis have not
yet been studied.
In the present study, human umbilical vein
endothelial cells (HUVECs) in suspension were used as a model for
anoikis. The cells in suspension or attached to culture plates were
treated with DHA. The cell death of HUVECs in these two models was
determined. Notably, 5 h treatment of 50 µM DHA significantly
increased the cell death of HUVECs in suspension, but not for
HUVECs attached to the plates. In addition, DHA specifically
activated the c-Jun N-terminal kinase (JNK) pathway in suspended
HUVECs, and the inhibition of the JNK pathway reversed the cell
death of HUVECs in suspension. These results suggest that DHA
promotes endothelial cell anoikis via the activation of the JNK
pathway.
Materials and methods
Cell culture
HUVECs were purchased from Lonza Group Ltd. (Basel,
Switzerland) and were cultured in endothelial basal cell medium-2
supplemented with EGM-2-MV bullet kit (Lonza Group Ltd.) and
antibiotics (100 international units/ml penicillin and 100 µg/ml
streptomycin). The cells were cultured at 37°C in a humidified
atmosphere containing 5% CO2. The HUVECs grown on
culture plates were used as attached cells. A group of confluent
cells were trypsinized for 2 min and detached to form single cell
suspension. These cells were cultured in suspension by slow
rotation in culture flasks and collected at 5 h as suspended
HUVECs. DHA (Sigma-Aldrich, St. Louis, MO, USA) and the JNK
inhibitor, SP600125 (Cell Signaling Technology, Inc., Danvers, MA,
USA), were dissolved in dimethyl sulfoxide (DMSO).
Trypan blue exclusion assay
Cell viability was assessed at 5 h after 50-µM DHA
treatment. The single cell suspensions were prepared and diluted
1:1 with 0.4% trypan blue (w/v in 0.9% NaCl; Santa Cruz
Biotechnology, Dallas, TX, USA). The dye-free cells were calculated
under a light microscope.
Flow cytometry
The cell death of HUVECs induced by DHA was detected
by using Annexin V-fluorescein isothiocyanate (FITC) and propidium
iodide (PI) staining (NeoBioscience, Shenzhen, China), according to
the manufacturer's instructions. Briefly, single cell suspensions
from attached or suspended HUVECs were prepared, washed with
phosphate-buffered saline (PBS) and resuspended in binding buffer
containing Annexin V-FITC (0.25%) and PI (1 µg/ml). An aliquot of
1×105 cells were examined using a FACSAria II flow
cytometer (BD Biosciences, San Jose, CA, USA). The percentages of
positive cells were analyzed using the FACSDiva version 6.0
acquisition and analysis software (BD Biosciences).
Western blotting
HUVECs were collected and washed with cold PBS, then
lysed in radioimmunoprecipitation assay buffer [20 mM Tris (pH
7.5), 150 mM NaCl, 50 mM NaF, 1% NP40, 0.1% deoxycholate, 0.1%
sodium dodecyl sulfate (SDS), 1 mM ethylenediaminetetraacetic acid,
1 mM phenylmethane sulfonyl fluoride and 1 mg/ml leupeptin].
Protein concentrations were determined using bicinchoninic acid
assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Equal
amounts of protein were separated by 8% SDS-polyacrylamide gel
electrophoresis and transferred to the polyvinylidene fluoride
(PVDF) membrane. After being blocked with 5% skimmed milk
overnight, the PVDF membranes were incubated with primary
antibodies in PBS-Tween at 4°C. The primary antibodies used were
rabbit monoclonal anti-phosphorylated- (p-)JNK (1:500; 4668; Cell
Signaling Technology, Inc.), rabbit polyclonal anti-JNK (1:500;
9252; Cell Signaling Technology, Inc.) and rabbit polyclonal
anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:5,000;
SAB2100894, Sigma-Aldrich). Immunoreactivity was visualized by
using a horseradish peroxidase-conjugated goat anti-rabbit
immunoglobulin G secondary antibody (1:2,000; 7074; Cell Signaling
Technology, Inc.) and a chemiluminescence kit (Pierce ECL; Thermo
Fisher Scientific, Inc., Waltham, MA USA). The densitometry
analyses were performed using ImageJ software (National Institutes
of Health, Bethesda, MD, USA). GAPDH levels were used as controls
for protein loading.
Statistical analyses
Each experiment was performed at least 3 times.
Statistical analyses were performed using SPSS version 11.5 (SPSS,
Inc., Chicago, IL, USA). The results are presented as the mean ±
standard deviation. A Student's t-test was used for statistical
comparisons between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
DHA inhibits the cell viability of
suspended endothelial cells
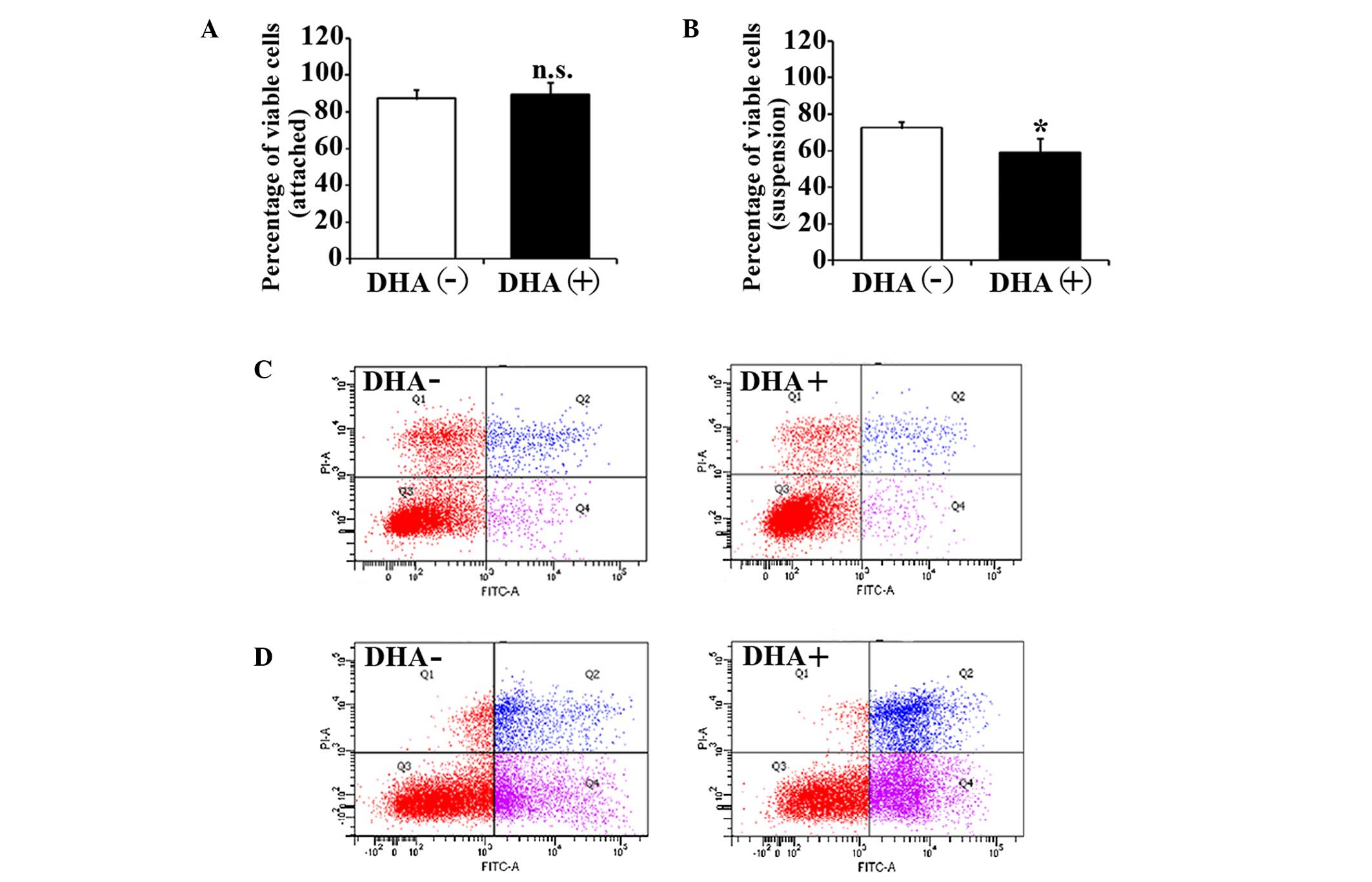
The effects of DHA on the cell viability of HUVECs
were evaluated by trypan blue exclusion assay. After 5 h in
suspension, the cell viability of HUVECs in the non-treatment group
was significantly decreased compared with the attached HUVECs
(88.3±4.6% vs. 73.6±3.1%; P=0.03). The percentage of viable cells
in attached HUVECs was not affected by 50 µM DHA after 5 h
incubation (88.3±4.6% vs. 90.7±6.1%; P=0.23) (Fig. 1A). However, the percentage of viable
cells in suspended HUVECs was significantly decreased after the
same DHA treatment (73.6±3.1% vs. 58.7±8.1%; P=0.02) (Fig. 1B). Therefore, DHA is likely to inhibit
the cell viability of suspended endothelial cells but not attached
endothelial cells.
DHA induces apoptosis in suspended
endothelial cells
To investigate the apoptotic status of the unviable
cells, HUVECs with DHA treatment were analyzed by flow cytometry
with Annexin V and PI staining. Consistent with viability assays,
increased apoptosis was observed in suspended HUVECs compared with
attached HUVECs in the non-treatment group (15.9±2.1% vs.
27.1±3.5%; P=0.04), suggesting that the suspended endothelial cells
undergo anoikis. DHA did not alter apoptosis in attached HUVECs
(15.9±2.1% vs. 16.3±1.7%; P=0.31) (Fig.
1C), but induced a significant increase of the apoptosis in
suspended HUVECs (27.1±3.5% vs. 39.3±4.4%; P=0.02) (Fig. 1D). This indicates that DHA
specifically enhances anoikis in endothelial cells.
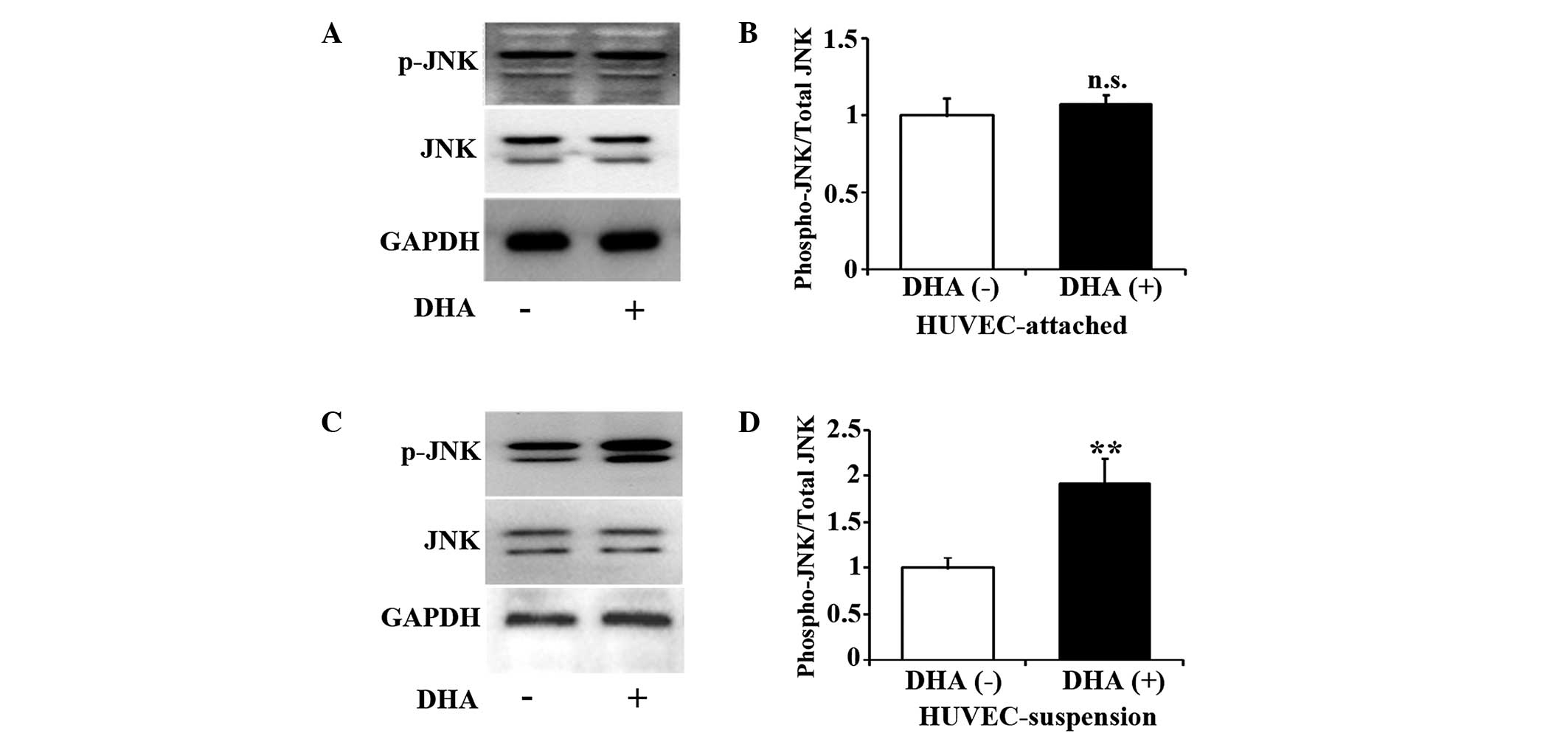
DHA activates the JNK pathway in
suspended endothelial cells
JNK is one of the major components of the
mitogen-activated protein kinase cascade, and participates in cell
death signaling pathways (17).
Studies have suggested that the JNK pathway may mediate anoikis in
epithelial cells (18,19). Therefore, the present study examined
the activation of JNK in DHA treated HUVECs by western blotting. As
shown in Fig. 2A and B, for up to 5 h
incubation with 50 µM DHA, the level of p-JNK remained unchanged in
attached HUVECs (P=0.13). However, the same treatment of DHA
significantly increased p-JNK in suspended HUVECs (P=0.01)
(Fig. 2C and D), suggesting that DHA
activates JNK pathway in suspended endothelial cells but not in
attached endothelial cells.
JNK inhibitor SP600125 reverses HUVEC
anoikis induced by DHA
SP600125 is a cell-permeable and selective inhibitor
of the JNK pathway (20). To further
verify the role of the JNK signaling pathway in the cell death of
suspended HUVECs, 10 µM SP600125 was applied to suspended HUVECs
for 1 h prior to DHA treatment. Fig.
3A indicates that SP600125 successfully prevented the increase
of p-JNK. SP600125 treatment abrogated the decrease of viable cells
in suspended HUVECs treated with DHA (74.6±4.5% vs. 78.9±6.6%;
P=0.19) (Fig. 3B). SP600125 also
attenuated DHA-induced apoptosis in suspended HUVECs (28.6±2.5% vs.
30.3±4.9%; P=0.12) (Fig. 3C). These
results suggest that JNK signaling pathway mediates DHA induced
anoikis in endothelial cells.
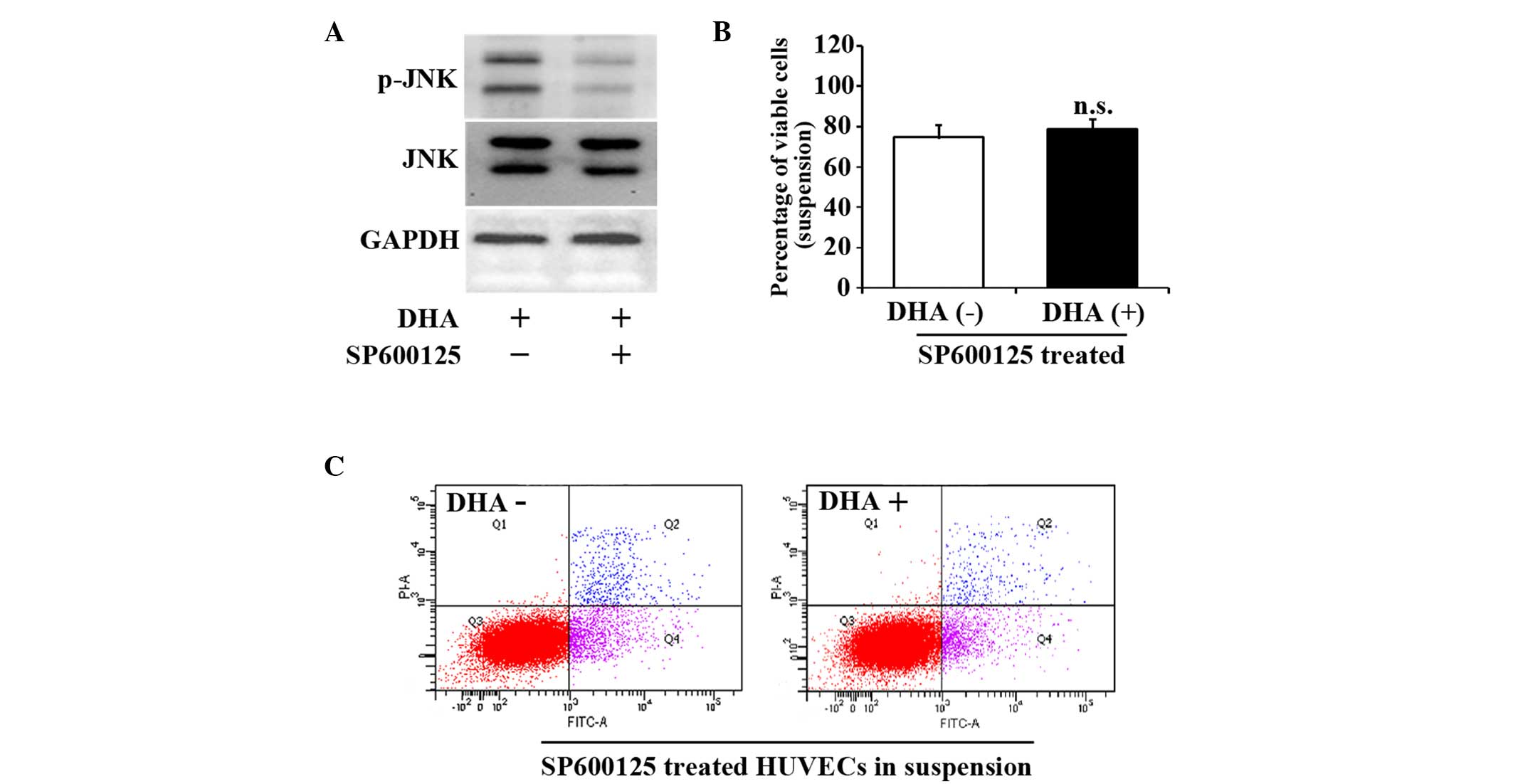 | Figure 3.JNK inhibitor, SP600125, reverses the
viability and apoptosis of suspended HUVECs induced by DHA. (A)
Representative immunoblots of p-JNK and JNK in suspended HUVECs
treated with DHA and SP600125. (B) Percentage of viable cells from
suspended HUVECs treated with DHA and SP600125; n=4. (C)
Representative images of flow cytometry analyses of Annexin
V/PI-staining in suspended HUVECs treated with DHA and SP600125.
DHA, dihydroartemisinin; JNK, c-Jun N-terminal kinase; p-JNK,
phosphorylated-JNK; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; HUVEC, human umbilical vein endothelial cell; n.s.,
non-significant; PI, propidium iodide; FITC, fluorescein
isothiocyanate. |
Discussion
DHA possesses strong anti-angiogenic activities, but
the molecular mechanisms are not yet fully understood (13,14,21). The
present study examined the effects of DHA on endothelial cell death
in attached and suspended HUVECs. After 5 h incubation, 50 µM DHA
induced cell death in suspended endothelial cells, but not in
attached endothelial cells. In addition, DHA increased the
expression of p-JNK, and blocking the JNK pathway abrogated
DHA-induced cell death in suspended endothelial cells. The present
study indicates that DHA induces endothelial cell anoikis via
activation of the JNK pathway.
Anoikis is induced by lack of correct cell or ECM
attachment, and anoikis resistance is important for tumor
metastasis (10). During tumor
angiogenesis, a group of endothelial cells migrate across the
basement membrane, leading to insufficient cell-matrix interactions
and subsequent anoikis (1).
Consistent with the findings of other reports, the present study
revealed the additional occurrence of cell death in untreated
endothelial cells in suspension, a model in which endothelial cells
are completely detached from the ECM. Endothelial cell anoikis is
crucial for tumor angiogenesis and presents a target for
anti-angiogenic therapy. In the current study, treatment with a low
concentration of DHA (50 µM) for a short-term exposure time (5 h)
was observed to induce cell death in suspended HUVECs. The same
treatment did not affect cell survival of attached HUVECs. This
suggests that DHA inhibits angiogenesis, which is at least
partially due to the induction of endothelial cell anoikis.
The JNK pathway, which appears to be activated by
detachment from the ECM, is critical for tumor cell apoptosis
(22). However, its role in anoikis
has been controversial. Several studies have reported that the
activation of the JNK signaling pathway mediates anoikis in cancer
cell lines (19,23,24). In
addition, B cell lymphoma-2 has been found to suppress the
suspension-induced activation of JNK signaling, which requires the
proteolytic function associated with interleukin-1-β-converting
enzyme (18). Conversely, the study
by Khwaja et al (25) reported
that the JNK pathway is not associated with anoikis in epithelial
cells. In the present study, DHA was demonstrated to promote
anoikis in suspended HUVECs through activation of the JNK signaling
pathway. In endothelial cells, detachment results in a rapid rise
in the level of reactive oxygen species (ROS), which modulate the
activity of the JNK signaling pathway (26). DHA increases the level of ROS in
several cancerous cell lines (27,28).
Another artemisinin derivative, artesunate, significantly inhibits
corneal neovascularization by inducing ROS-dependent apoptosis in
vascular endothelial cells (29).
Therefore, DHA may induce an increase in ROS and then activate then
JNK signaling pathway in suspended HUVECs.
The JNK pathway may also interact with focal
adhesion kinase (FAK) signaling in mediating anoikis. FAK is a key
component of cell-substratum adhesions, and disruption of FAK
signaling results in a loss of substrate adhesion and anoikis in
endothelial cells (30).
Glucocorticoids induce osteocyte anoikis by blocking FAK signaling
and activating JNK. In addition, FAK blocks the ras-related C3
botulinum toxin substrate 1/JNK pathway in vascular smooth muscle
cells (31). However, in lung
adenocarcinoma cells, FAK regulates anoikis independently of the
JNK pathway (32). DHA directly
decreases the level of p-FAK in ovarian cancer cells (33). Thus, the role of FAK signaling in DHA
induced anoikis requires further investigation.
In summary, the present study demonstrates that DHA
induces endothelial cell anoikis, which is mediated by the
activation of the JNK pathway. DHA may be considered as a promising
angiogenesis inhibitor for clinical application. The findings of
the present study will aid the current understanding of the
molecular mechanisms underlying the anti-angiogenic effects of
DHA.
Acknowledgements
The present study was supported by the Medical
Science and Technology Development Plan of Shandong Province
(Jinan, China; grant no. 2013WS0137). The authors are grateful for
the support provided to Professor Ju Liu from the Shandong Taishan
Scholarship (Jinan, China).
References
|
1
|
Kerbel RS: Tumor angiogenesis. N Engl J
Med. 358:2039–2049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Folkman J: Angiogenesis in cancer,
vascular, rheumatoid and other disease. Nat Med. 1:27–31. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carmeliet P: Mechanisms of angiogenesis
and arteriogenesis. Nat Med. 6:389–395. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Folkman J: Tumor angiogenesis: Therapeutic
implications. N Engl J Med. 285:1182–1186. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu J, Deutsch U, Jeong J and Lobe CG:
Constitutive notch signaling in adult transgenic mice inhibits
bFGF-induced angiogenesis and blocks ovarian follicle development.
Genesis. 52:809–816. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim KJ, Li B, Winer J, Armanini M, Gillett
N, Phillips HS and Ferrara N: Inhibition of vascular endothelial
growth factor-induced angiogenesis suppresses tumour growth in
vivo. Nature. 362:841–844. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hanahan D: Signaling vascular
morphogenesis and maintenance. Science. 277:48–50. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aoudjit F and Vuori K: Matrix attachment
regulates Fas-induced apoptosis in endothelial cells: A role for
c-flip and implications for anoikis. J Cell Biol. 152:633–643.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Green DR and Llambi F: Cell death
signaling. Cold Spring Harb Perspect Biol. 7:pii: a006080. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gilmore AP: Anoikis. Cell Death Differ.
12(Suppl 2): 1473–1477. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee J, Zhou HJ and Wu XH:
Dihydroartemisinin downregulates vascular endothelial growth factor
expression and induces apoptosis in chronic myeloid leukemia K562
cells. Cancer Chemother Pharmacol. 57:213–220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ho WE, Peh HY, Chan TK and Wong WS:
Artemisinins: Pharmacological actions beyond anti-malarial.
Pharmacol Ther. 142:126–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong F, Zhou X, Li C, Yan S, Deng X, Cao
Z, Li L, Tang B, Allen TD and Liu J: Dihydroartemisinin targets
VEGFR2 via the NF-κB pathway in endothelial cells to inhibit
angiogenesis. Cancer Biol Ther. 15:1479–1488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dong F, Tian H, Yan S, Li L, Dong X, Wang
F, Li J, Li C, Cao Z, Liu X and Liu J: Dihydroartemisinin inhibits
endothelial cell proliferation through the suppression of the ERK
signaling pathway. Int J Mol Med. 35:1381–1387. 2015.PubMed/NCBI
|
|
15
|
Guo L, Dong F, Hou Y, Cai W, Zhou X, Huang
AL, Yang M, Allen TD and Liu J: Dihydroartemisinin inhibits
vascular endothelial growth factor-induced endothelial cell
migration by a p38 mitogen-activated protein kinase-independent
pathway. Exp Ther Med. 8:1707–1712. 2014.PubMed/NCBI
|
|
16
|
Chen HH, Zhou HJ, Wang WQ and Wu GD:
Antimalarial dihydroartemisinin also inhibits angiogenesis. Cancer
Chemother Pharmacol. 53:423–432. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu J and Kapron CM: Differential
induction of MAP kinase signalling pathways by cadmium in primary
cultures of mouse embryo limb bud cells. Reprod Toxicol.
29:286–291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Frisch SM, Vuori K, Kelaita D and Sicks S:
A role for Jun-N-terminal kinase in anoikis; suppression by bcl-2
and crmA. J Cell Biol. 135:1377–1382. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cardone MH, Salvesen GS, Widmann C,
Johnson G and Frisch SM: The regulation of anoikis: MEKK-1
activation requires cleavage by caspases. Cell. 90:315–323. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oh S, Jeong IH, Shin WS and Lee S: Growth
inhibition activity of thioacetal artemisinin derivatives against
human umbilical vein endothelial cells. Bioorg Med Chem Lett.
13:3665–3668. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leppa S and Bohmann D: Diverse functions
of JNK signaling and c-Jun in stress response and apoptosis.
Oncogene. 18:6158–6162. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Rivera Rosado LA, Moon SY and
Zhang B: Silencing of D4-GDI inhibits growth and invasive behavior
in MDA-MB-231 cells by activation of Rac-dependent p38 and JNK
signaling. J Biol Chem. 284:12956–12965. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Krestow JK, Rak J, Filmus J and Kerbel RS:
Functional dissociation of anoikis-like cell death and activity of
stress activated protein kinase. Biochem Biophys Res Commun.
260:48–53. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Khwaja A and Downward J: Lack of
correlation between activation of Jun-NH2-terminal kinase and
induction of apoptosis after detachment of epithelial cells. J Cell
Biol. 139:1017–1023. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li AE, Ito H, Rovira II, Kim KS, Takeda K,
Yu ZY, Ferrans VJ and Finkel T: A role for reactive oxygen species
in endothelial cell anoikis. Circ Res. 85:304–310. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kong R, Jia G, Cheng ZX, Wang YW, Mu M,
Wang SJ, Pan SH, Gao Y, Jiang HC, Dong DL and Sun B:
Dihydroartemisinin enhances Apo2L/TRAIL-mediated apoptosis in
pancreatic cancer cells via ROS-mediated up-regulation of death
receptor 5. PloS One. 7:e372222012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim SJ, Kim MS, Lee JW, Lee CH, Yoo H,
Shin SH, Park MJ and Lee SH: Dihydroartemisinin enhances
radiosensitivity of human glioma cells in vitro. J Cancer
Res Clin Oncol. 132:129–135. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng R, Li C, Wei L, Li L, Zhang Y, Yao
Y, Gu X, Cai W, Yang Z, Ma J, et al: The artemisinin derivative
artesunate inhibits corneal neovascularization by inducing
ROS-dependent apoptosis in vascular endothelial cells. Invest
Ophthalmol Vis Sci. 54:3400–3409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu Q and Rounds S: Focal adhesion kinase
and endothelial cell apoptosis. Microvasc Res. 83:56–63. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sundberg LJ, Galante LM, Bill HM, Mack CP
and Taylor JM: An endogenous inhibitor of focal adhesion kinase
blocks Rac1/JNK but not Ras/ERK-dependent signaling in vascular
smooth muscle cells. J Biol Chem. 278:29783–29791. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu G, Meng X, Jin Y, Bai J, Zhao Y, Cui
X, Chen F and Fu S: Inhibitory role of focal adhesion kinase on
anoikis in the lung cancer cell A549. Cell Biol Int. 32:663–670.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu B, Hu K, Li S, Zhu J, Gu L, Shen H,
Hambly BD, Bao S and Di W: Dihydroartiminisin inhibits the growth
and metastasis of epithelial ovarian cancer. Oncol Rep. 27:101–108.
2012.PubMed/NCBI
|