Introduction
In 1968, Steiner et al first used the term
MEN2, which is now known as multiple endocrine neoplasia 2A (MEN2A)
(1). In 1986, Farndon et al
decribed familial medullary thyroid carcinoma (MTC), which appears
to be closely associated with MEN2A (2). The phenotypes of MEN2A include bilateral
multiple MTC, unilateral or bilateral phaeochromocytoma (PHEO), and
parathyroid adenoma or hyperplasia (3). In the majority of cases, the diagnosis
of PHEO is determined following the diagnosis of MTC. The first
symptom of the patient in the present study was bilateral PHEO,
with a phenotype of parathyroid adenoma. However, the
manifestations of MEN2A indicated in the patient's son were of MTC
and PHEO, despite the same genotype being present. However, no
occurrence of additional mutations was present. These findings
inferred that there may be another proto-oncogene dysfunction or
activation present, in addition to the multiple superimposed
effects of mutation epigenetics. The interaction between these
genetic factors and environmental effects may have lead to the
varied clinical manifestations.
Case report
A 73-year-old man presented to The First Affiliated
Hospital of China Medical University (Shenyang, China) in july 2013
with persistently increased levels of serum calcium. The patient
had undergone a bilateral adrenalectomy for PHEO 4 years
previously. An ultrasound examination of the thyroid gland revealed
a low echo, 16.0×10.4-mm nodule, in addition to the elevated levels
of parathyroid hormone (81.06 pmol/l; normal range, 0.66–12
pmol/l), serum calcitonin (53.79 pmo/l; normal range, 0.58–22
pmol/l) and serum carcinoembryonic antigen (CEA; 7.24 ng/ml; normal
range, 0.0–4.3 ng/ml) indicated by a laboratory examination. A
bilateral adrenal ultrasound revealed kidney stones in the left
kidney and a contrast-enhanced cervical computed tomography (CT)
scan showed a well-defined enhancing lesion in the right thyroid
and a lesion in the parathyroid, which suggested the diagnosis of
thyroid and parathyroid adenomas. The genomic DNA of the proband
was extracted from peripheral blood using a DNA extraction and
purification kit (Qiagen GmbH, Hilden, Germany). Shenzhen Huada
Gene Technology Co., Ltd., (Shenzhen, China) was authorized to
amplify all exons of the rearrangement during transfection (RET)
and menin genes, then perform bidirectional sequencing. The
heterozygous polymorphism, TGC→CGC, at codon 634 (C634R) on the
exon 11 rs377767437 sequence of the RET proto-oncogene was detected
in the patient, which resulted from the C634R cysteine to arginine
substitution. The patient underwent a parathyroid adenoma resection
in June 2014 and the diagnosis of parathyroid adenoma was confirmed
by histological examination (Fig.
1A), however, the patient refused treatment for the thyroid
lesion. The patient was lost to follow-up.
The 43-year-old son of the patient underwent genetic
testing, which indicated heterozygosity for the same mutation at
C634R. The symptoms of the son included bilateral PHEO, calcitonin
levels of >1,450 pmol/l, elevated CEA levels 43.6 ng/ml and
normal parathyroid hormone levels. Repeated 24-h urinary tests
showed elevated levels of catecholamines, at 1,530.2 mol/24 h
(normal range, 59.1–266 mol/24 h). MTC was diagnosed from the
thyroid ultrasound, the elevated calcitonin and CEA levels, and the
post-operative pathology (Fig. 1B-D).
The post-operative pathology of the patient's son was composed of
polygonal or plump spindle cells. Characteristic slender
fibrovascular septas were present in the carcinoma mass. Island- or
rosette-like cell clusters and stromal amyloid deposition were also
present. Immunohistochemical analysis revealed medullary carcinoma,
with positive expression of calcitonin and thyroid transcription
factor-1. Following surgery, the levels of parathyroid hormone,
serum calcitonin and serum CEA were markedly decreased, but still
above the standard range. The level of parathyroid hormone was
14.37 pmol/l, the level of serum calcitonin was 416.4 pmol/l and
the level of serum CEA was 13.73 ng/ml.
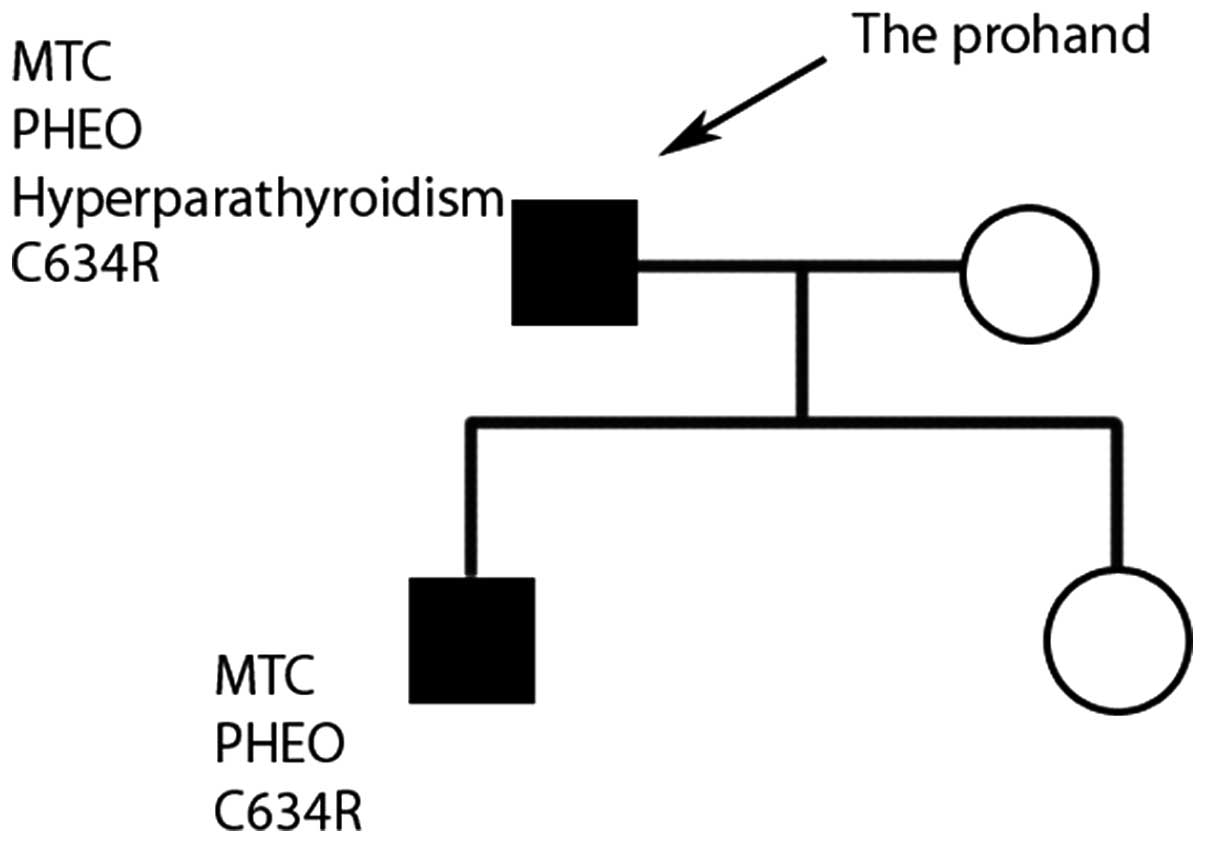
The wife and daughter of the patient did not possess
enlarged neck masses or the manifestations of hypocalcemia. No
associated mutations were identified during genetic screening
(Fig. 2).
Discussion
MEN2A is a genetic dominant syndrome resulting from
specific RET proto-oncogene mutations, which is characterized by
the coexistence of adrenal PHEO, MET and hyperparathyroidism
(4) Numerous RET gene mutational
sites have been previously reported, 95% of which are located at 5
coding Cys codons: Codons 609, 611, 618 and 620 of exon 10 and
C634R of exon 11. All mutations of codon 634 constitute 73–85% of
RET mutations. The C634Y mutation is the most commonly occurring of
the codon 634 mutations, followed by the C634Y mutation in Chinese
individuals (5). PHEO mainly occurs
in association with codon 634 and 918 mutations, but rarely in
association with codon 618, codon 620 or other mutations (6–8). A
previous study reported that the majority of patients with a codon
634 mutation will develop PHEO and MTC during their lifetime
(9), as occurred in the case in the
present study. A patient with a codon 634 mutation is prone to
develop PHEO and MTC at the same time. The diagnosis of PHEO may be
determined prior to MTC (12.9–25.1% of patients), simultaneously
with MTC (34.7–38.9% of patients) or following the diagnosis of MTC
(40.2–48.2% of patients) (10). In
addition, half of all patients with PHEO may be asymptomatic at
diagnosis (9,10). However, the patient and son in the
present study were diagnosed with labile hypertension, which is the
typical manifestation of PHEO. Patients with typical MTC symptoms
present with thyromegaly and pain in the front of the neck prior to
the age of 35 years. The symptoms of patients with MTC and C634R
mutations are more severe compared with patients only diagnosed
with MTC (7). Recently, the American
Thyroid Association classified the risk levels of MEN2A, according
to all recognized mutations and the aggressive nature of codon 634
(11). The patient in the present
study had no evident nodules or neck nodes, or other characteristic
manifestations of MTC at the first visit to the hospital.
Therefore, the first symptom of the patient was inferred to most
probably be PHEO.
Previous studies have reported that the C634R
mutation constitutes 54% of all diseases caused by mutations in the
MEN2A family. Additionally, 65% of all changes to codon 634 and
MEN2A patients with a C634R substitution confer an increased risk
of developing parathyroid disease compared with other mutations in
codon 634 (12). In the present
study, the patient was diagnosed with hyperplasia in the
parathyroid gland; however, no symptoms or biochemical examination
results that are associated with hyperplasia in the parathyroid
gland were exhibited by the patient's son, which lead to the
hypothesis that there may be a variety of mutations occurring in
patients from the same family. In 2001, Mathew et al
reported the case of an 18-year-old MEN2A patient with MTC, who
succumbed due to multiple metastases. In addition to the C634R
mutation, the loss of heterozygosity (retLOH) in exon 4-exon 16 is
found in metastasis (13). In 1999,
Tessitore et al reported the case of another MEN2A patient
who possessed C634R and A640G double mutations (14). Double RET mutations may cause unusual
MEN2 manifestations. The ‘second hit’-superposition of two
variations may be the reason why members of the same family exhibit
various phenotypes (15). In the
present study, however, the two patients possessed no other
mutations. Therefore, in addition to the multiple superimposed
effects of mutation, there may be another dysfunction or activation
of the proto-oncogene present. The interaction between genetic
factors and environmental effects may have lead to the varied
clinical manifestations possessed by the patient and his son.
The RET gene mutation has a high specificity and the
penetrance of the mutation is almost 100%, which is helpful for the
early diagnosis of the disease (16).
The current guidelines recommend a DNA analysis of the patient and
kin for RET proto-oncogene mutations (6). All individuals with the RET gene
mutation require a prophylactic thyroidectomy, and the timing of
the surgery is associated with the presence of specific mutations
(17). Certain studies consider the
sequencing of the RET gene to investigate germline mutations as the
standard screening test for MEN2 syndromes (18). The patient in the present study
possessed the typical manifestations, biological results and
morphological features of MEN2A; however, the patient's son did not
exhibit all of these. Therefore, a family history, biological
examinations and DNA screening are of high importance in order to
ensure the correct diagnosis.
The patient and son were diagnosed with definite
MEN2A. The molecular genetic basis of the disease is the 17th base
T→C mutation in the exon 11 rs377767437 sequence. Another
proto-oncogene dysfunction or activation in interaction with
environmental factor epigenetics may have caused the varying
clinical manifestations between father and son.
References
|
1
|
Steiner AL, Goodman AD and Powers SR:
Study of a kindred with pheochromocytoma, medullary thyroid
carcinoma, hyperparathyroidism and Cushing's disease: Multiple
endocrine neoplasia, type 2. Medicine (Baltimore). 47:371–409.
1968. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Farndon JR, Leight GS, Dilley WG, Baylin
SB, Smallridge RC, Harrison TS and Wells SA: Familial medullary
thyroid carcinoma without associated endocrinopathies: A distinct
clinical entity. Br J Surg. 73:278–281. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ezzat T, Paramesawaran R, Phillips B and
Sadler G: MEN 2 syndrome masquerading as MEN 1. Ann R Coll Surg
Engl. 94:e206–e207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Muzannara MA, Tawfeeq N, Nasir M, Al Harbi
MK, Geldhof G and Dimitriou V: Vaginal delivery in a patient with
pheochromocytoma, medullary thyroid cancer, and primary
hyperparathyroidism (multiple endocrine neoplasia type 2A, Sipple's
syndrome). Saudi J Anaesth. 8:437–439. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lau GS, Lang BH, Lo CY, Tso A,
Garcia-Barcelo MM, Tam PK and Lam KS: Prophylactic thyroidectomy in
ethnic Chinese patients with multiple endocrine neoplasia type 2A
syndrome after the introduction of genetic testing. Hong Kong Med
J. 15:326–331. 2009.PubMed/NCBI
|
|
6
|
American Thyroid Association Guidelines
Task Force. Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib
H, Moley JF, Pacini F, Ringel MD, Schlumberger M and Wells SA Jr:
Medullary thyroid cancer: Management guidelines of the American
Thyroid Association. Thyroid. 19:565–612. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brandi ML, Gagel RF, Angeli A, Bilezikian
JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG,
Libroia A, et al: Guidelines for diagnosis and therapy of MEN type
1 and type 2. J Clin Endocrinol Metab. 86:5658–5671. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moley JF: The molecular genetics of
multiple endocrine neoplasia type 2A and related syndromes. Annu
Rev Med. 48:409–420. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Imai T, Uchino S, Okamoto T, Suzuki S,
Kosugi S, Kikumori T and Sakurai A: MEN Consortium of Japan: High
penetrance of pheochromocytoma in multiple endocrine neoplasia 2
caused by germ line RET codon 634 mutation in Japanese patients.
Eur J endocrinol. 168:683–687. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rowland KJ, Chernock RD and Moley JF:
Pheochromocytoma in an 8-year-old patient with multiple endocrine
neoplasia type 2A: Implications for screening. J Surg Oncol.
108:203–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Krampitz GW and Norton JA: RET gene
mutations (genotype and phenotype) of multiple endocrine neoplasia
type 2 and familial medullary thyroid carcinoma. Cancer.
120:1920–1931. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mulligan LM, Kwok JB, Healey CS, Elsdon
MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, et al:
Germ-line mutations of the RET proto-oncogene in multiple endocrine
neoplasia type 2A. Nature. 363:458–460. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mathew CG, Smith BA, Thorpe K, Wong Z,
Royle NJ, Jeffreys AJ and Ponder BA: Deletion of genes on
chromosome 1 in endocrine neoplasia. Nature. 328:524–526. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tessitore A, Sinisi AA, Pasquali D,
Cardone M, Vitale D, Bellastella A and Colantuoni V: A novel case
of multiple endocrine neoplasia type 2A associated with two de novo
mutations of the RET protooncogene. J Clin Endocrinol Metab.
84:3522–3527. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qi X-P, Ma J-M, Du Z-F, Ying R-B, Fei J,
Jin HY, Han JS, Wang JQ, Chen XL, Chen CY, et al: RET germline
mutations identified by exome sequencing in a Chinese multiple
endocrine neoplasia type 2A/familial medullary thyroid carcinoma
family. PLoS One. 6:e203532011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gosnell JE, Sywak MS, Sidhu SB, Gough IR,
Learoyd DL, Robinson BG and Delbridge LW: New era: Prophylactic
surgery for patients with multiple endocrine neoplasia-2a. ANZ J
Surg. 76:586–590. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Balachandran K, Kamalanathan S,
Gopalakrishnan S and Murugananadham K: Multiple endocrine neoplasia
2B: Delayed presentation, rapid diagnosis. BMJ Case Rep.
2013:bcr20130091852013.PubMed/NCBI
|
|
18
|
Cui Q, Wang W, Fu Z, Shao X, Zhang Z,
Zhang M, Ju X, Wang K, Chen J and Zhou H: Integrated
DNA-based/biochemical screening for early diagnosis of multiple
endocrine neoplasia type 2A (MEN2A). J Biomed Res. 27:145–150.
2013. View Article : Google Scholar : PubMed/NCBI
|