Introduction
Testicular cancer (TC) is a frequently occurring
disease among adult males (1). TC
develops in the testicles and may include various types of cancer,
including germ cell tumors (GCT), sex cord or gonadal stromal
tumors, and secondary tumors of the testes (1). Over 95% of TC cases are testicular GCTs
(TGCTs) (2). Although TC has the
highest cure rate of all forms of cancer and a 5-year survival rate
of >90% (1), TC remains to be the
most common cancer in young to middle-aged men (3).
MicroRNAs (miRNAs/miRs) and transcription factors
(TFs) are key regulators of gene expression (4). TFs are specific proteins that control
transcription by regulating gene expression at the transcriptional
level, either working alone or with other proteins. TFs function to
activate or repress transcription by binding to specific DNA
sequences, known as cis-regulatory elements, located in the
upstream regions of genes, whilst miRNAs are small, non-coding RNA
molecules that affect gene expression at the post-transcription
level (4).
miRNAs target genes that are associated with, and
are important in, a number of biological processes, including
apoptosis, differentiation and proliferation. Studying these target
genes (targets) offers an intuitive method to identify the effect
of miRNAs at the transcriptional level. Genes that contain miRNAs
are known as host genes (hosts). Rodriguez et al (5) reported that miRNAs and host transcripts
are transcribed in parallel, and identified two transcription
classes of miRNAs, known as intronic and exonic. Baskerville and
Bartel (6) demonstrated that there is
a close association between host genes and their intronic miRNA,
which are often expressed together, forming a specific association
that allows them to achieve full biological function and
subsequently modify pathways (7).
miRNAs and differentially-expressed genes involved
in TC have been investigated for >10 years in this century.
However, studies at the gene level are currently only based on a
single aspect, either genes or miRNAs. As a result, identifying the
genes of interest in the pathogenesis of TC is challenging. The
present study aimed to take a more global perspective, determining
the underlying networks of miRNAs, TFs, targets and hosts to
analyze the basic gene pathogenesis. The results identified three
types of association: miRNAs target genes, and TFs regulate miRNAs
and miRNA host genes. The global network is necessary for research,
but cannot be used easily to extract the key regulatory elements;
therefore, differentially-expressed and related networks were also
determined. The latter two networks were used specifically to
extract key pathways sustained by differentially-expressed
elements. To compare the similarities and differences between these
pathways, the current study identified key elements at the gene
level of TC regulation. The results of the present study reveal the
regulatory associations of TC and are important in further aiding
the research in this field. If mutated factors can be regulated and
maintained within normal ranges, the development of TC may be
prevented.
Materials and methods
Material collection and data
processing
Firstly, the present study collected experimentally
validated datasets of human TC miRNAs and their target genes from
Tarbase 5.0 (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index)
and miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/). The symbols and
names obtained were normalized using the official symbols and names
from the National Center for Biotechnology Information (NCBI)
database (http://www.ncbi.nlm.nih.gov/gene/). Different forms of
TC were not classified during the data collection process. The
majority of data collected referred to TGCT, which is the most
common TC subtype.
Secondly, the current study obtained a human
experimentally validated dataset of TFs and miRNAs from TransmiR
(8), which collects and summarizes
data from public literatures and biological experiments.
Thirdly, human TC host genes were extracted from
miRBase (9) and the NCBI database,
and each host gene was assigned an official symbol and
identification code.
Fourthly, the differentially-expressed genes
obtained in the present study were extracted from Cancer Genetics
Web (http://www.cancerindex.org/geneweb/), NCBI, and
relative literatures and papers. The differentially-expressed
elements that were extracted included abnormally expressed
proteins, genetic mutations, single nucleotide polymorphisms, and
upregulated, downregulated, overexpressed and variable genes. The
current study extracted TC-related genes from the GeneCards
database (10) and relevant
literature. TC-related genes affect the growth and migration of
tumors, in addition to the efficacy of radial therapy and the
clinical outcome of TC (11).
Furthermore, popular TFs were obtained using the P-match method
(12). All results were combined to
produce a list of the TC relative genes. The present study only
focused on the TFs obtained using TransmiR. The 1000-nucleotide
(nt) promoter region sequences of the targets of
differentially-expressed genes were downloaded from the University
of California, Santa Cruz database (13). The P-match method was used in
combination with weight matrix and pattern matching approaches to
identify transcription factor binding sites (TFBSs) in the 1000-nt
promoter region sequences, and the TFBSs were subsequently mapped
onto the promoter regions of targets. The P-match matrix library
(http://www.gene-regulation.com/cgi-bin/pub/programs/pmatch/bin/p-match.cgi),
along with information collected from TRANSFAC®
(http://www.gene-regulation.com/pub/databases.html),
contained sets of known TFBSs, and were used to search for a large
number of different TFBSs. The vertebrate matrix and restricted
high quality criterion of the matrix were used in particular for
the search.
Finally, differentially-expressed miRNAs were
extracted from miR2Disease (14) and
relevant literatures. miR2Disease is a database that records miRNAs
that are differentially-expressed in a number of human diseases.
Novel experimental data was also obtained manually from recent
studies and science citation index papers (http://apps.webofknowledge.com) regarding TC
differentially-expressed miRNAs, using the key words ‘seminoma’,
‘testicular germ cell tumor’ and ‘testicular cancer’.
Network construction
Data was organized into tables and subsequently used
to form a graph. The TC-related factors, including TFs, genes and
miRNAs, form the nodes, and the lines placed between these nodes
signify the form of association between each element. As a result,
the associations between elements form regulatory networks.
Three types of TC regulatory networks were formed,
including the i) differentially-expressed network, ii) related
network and iii) global network. Regulatory associations were
extracted for TFs, miRNAs, and target and host genes from the
dataset collected by the present study. This formed the global
regulatory network, which combined all associations.
Differentially-expressed elements were determined using the
aforementioned fourth and final steps, and the associations between
them, which were obtained from the global network, were selected to
derive the differentially-expressed network. The related network
was formed in a similar way.
Results
Differentially-expressed network of
TC
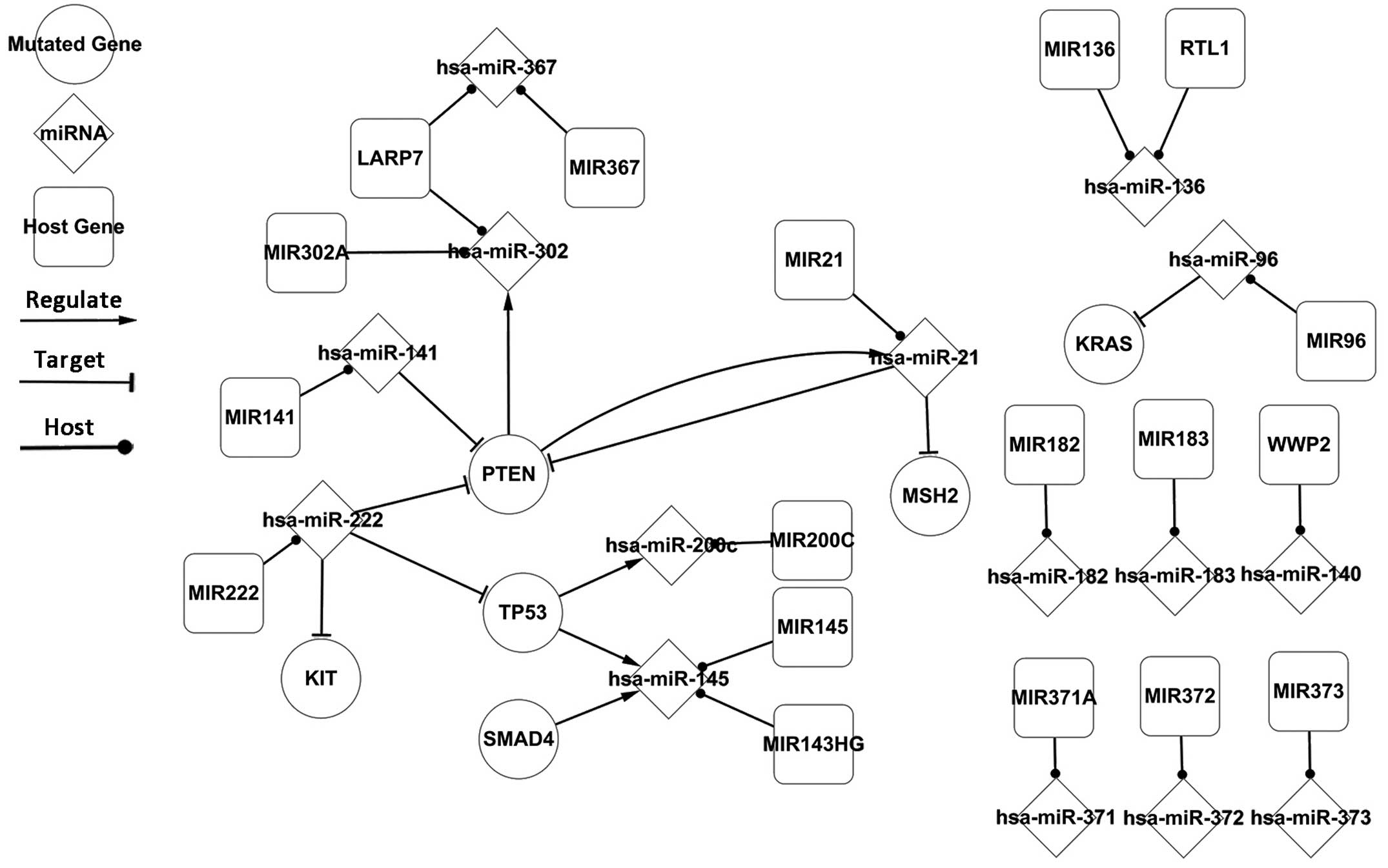
Fig. 1 presents the
regulatory associations of all differentially-expressed elements in
TC. It is composed of TFs, miRNAs, and targets and hosts of miRNAs.
This network was key for the latter half of the present study. The
nodes in the network impact others directly or indirectly, and when
combined, the associations reveal the primary cause of TC. If the
genes and miRNAs are not differentially-expressed as presented in
the network, then that individual is not expected to develop TC.
Thus, TC may be prevented through the suppression of these
differentially-expressed elements.
Similar to the host genes, all nodes are
differentially-expressed in TC. The network contains three TFs:
Tumor protein 53 (TP53), SMAD family member 4 (SMAD4) and
phosphatase and tensin homolog (PTEN). The most significant
pathways contain these three TFs. As presented in Fig. 1, TP53 and PTEN are together targeted
by hsa-miR-222. TP53 is a common, mutated TF, which is
differentially-expressed in various types of cancer (15), and the present study identified that
it regulates hsa-miR-200c and hsa-miR-145 (Fig. 1). SMAD4 is a novel transcription
factor, which is mutated in seminomatous GCTs (16), and the current study determined that
it regulates hsa-miR-145 together with TP53. PTEN is an important
junction of the network (17). PTEN
and hsa-miR-21 form a self-adaptation association, forming the only
circuit in the whole network (Fig.
1). The combined action of the nodes in the
differentially-expressed network partially exposes the regulatory
mechanisms of TC. The network indicates that it may be possible to
artificially regulate differentially-expressed elements back to
normal levels as a method to prevent or cure TC.
Related network of TC
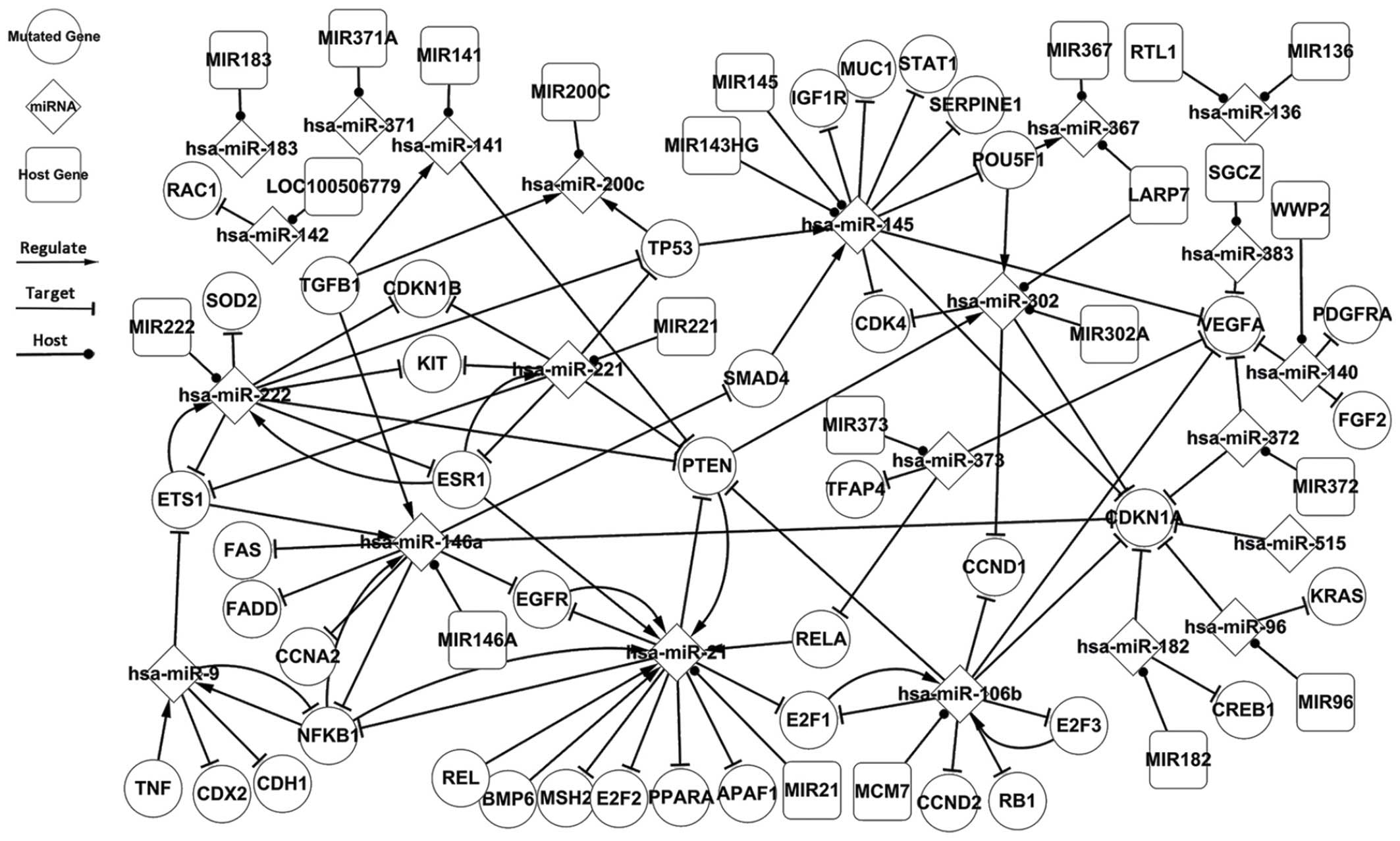
Fig. 2 presents the
mass regulatory associations between genes and miRNAs in TC. The
related network includes the related elements collected by the
present study, in addition to predicted data. It is evident that
the related network includes the differentially-expressed network.
The related network of TC includes 18 TFs, 26 miRNAs and a number
of targets. Fig. 2 presents certain
integral, regulatory associations beyond the
differentially-expressed network with a few additional pathways.
The miRNA, hsa-miR-21, is regulated by 7 TFs and forms 3
self-adaptation associations. The TF, SMAD4, is targeted by
hsa-miR-146a and regulates only 1 miRNA, hsa-miR-145. Other TF
associations may also be identified within the related network,
including TGFB1, which regulates 3 miRNAs, but is not targeted by
other miRNAs, and ESR1, which regulates hsa-miR-21 and forms 2
self-adaptation associations with hsa-miR-221 and hsa-miR-222. The
related network expands the additional topological associations to
the pathogenesis of TC, and includes elements that are not
differentially-expressed.
Global network of TC
The global network presents a greater number of
comprehensive, regulatory associations compared with the
differentially-expressed and related networks. The global network
involves the whole experimentally validated network of the human
body, and incorporates the related and differentially-expressed
networks. All regulation elements and associations are common
regulation situations that are present not only in TC, but in other
diseases.
Host genes and miRNAs in TC
Host genes are not differentially-expressed in
cancer, but are considered to be differentially-expressed if their
miRNAs are. For example, la ribonucleoprotein domain family, member
7 hosts two differentially-expressed miRNAs, hsa-miR-367 and
hsa-miR-302, which are each regulated by POU class 5 homeobox 1
(POU5F1). POU5F1 is targeted by hsa-miR-145, which is
differentially-expressed, suggesting that the host genes may be
useful in the regulatory pathway. Certain host genes contain a
number of miRNAs that target genes or are regulated by other
important TFs. The host genes and their differentially-expressed
miRNAs may aid the understanding of TC pathogenesis.
Regulatory pathways of
differentially-expressed genes
Differentially-expressed genes are presented in the
differentially-expressed network. These genes directly regulate
various differentially-expressed miRNAs, and therefore may be
considered, to a certain extent, as a source of differential
expression. Adjusting the differentially-expressed TFs to their
normal condition may be a possible method for altering the genes
associated with TC, via miRNAs, which would be affected the at the
same time.
In order to clearly describe the features of the TC
networks, the up- and downstream information of genes or miRNAs was
extracted and presented in tables. Table
I lists the successors and precursors of PTEN as determined by
the related, differentially-expressed and global networks. PTEN is
differentially-expressed itself and associated with other
differentially-expressed genes or miRNAs in the network. PTEN and
hsa-miR-21 form a self-adaptation association, thus affecting other
genes as a unit. There are two miRNAs downstream of PTEN in the
related network, which are each differentially-expressed. The
expression of PTEN may be controlled as a source to adjust the
differentially-expressed miRNAs that are regulated by PTEN. By
controlling the expression of specific genes in the network, the
abnormal expression of other elements may also be controlled,
resulting in TC prevention.
 | Table I.Upstream and downstream miRNAs of the
PTEN gene in testicular cancer. |
Table I.
Upstream and downstream miRNAs of the
PTEN gene in testicular cancer.
| Location | Global network | Related network |
Differentially-expressed network |
|---|
| PTEN |
|
|
|
|
Upstream | hsa-miR-106b,
hsa-miR-141, hsa-miR-17, hsa-miR-18a, hsa-miR-19a, hsa-miR-19b,
hsa-miR-20a, hsa-miR-21, hsa-miR-214, hsa-miR-216a, hsa-miR-217,
hsa-miR-221, hsa-miR-222, hsa-miR-26a, hsa-miR-494 | hsa-miR-106b,
hsa-miR-141, hsa-miR-21, hsa-miR-221, hsa-miR-222 | hsa-miR-141,
hsa-miR-21 hsa-miR-222 |
|
Downstream | hsa-miR-19a,
hsa-miR-21, hsa-miR-21, hsa-miR-22, hsa-miR-25, hsa-miR-302a,
hsa-miR-302b, hsa-miR-302c, hsa-miR-302d, hsa-miR-302f | hsa-miR-21,
hsa-miR-302 | hsa-miR-21,
hsa-miR-302 |
Regulatory pathways of
differentially-expressed miRNAs
Differentially-expressed miRNAs are important in the
transcription network due to their association with other
associated genetic elements, and therefore warrant further
investigation. Theoretically, differentially-expressed miRNAs may
be artificially regulated indirectly through regulating or
targeting associations. Therefore, the present study extracted,
compared and analyzed these miRNAs using the same method applied to
analyze the differentially-expressed genes.
hsa-miR-21 is presented as an example in Table II. The data indicated that hsa-miR-21
is important in the transcription network. hsa-miR-21 is regulated
by 7 genes, which are all TFs, and targets 7 genes, which include
three TFs [epidermal growth factor receptor (EGFR), E2F
transcription factor 1 (E2F1) and PTEN]. PTEN and hsa-miR-21 form
an important self-adaptation association, which was discussed
previously in the current study. Similarly, 2 self-adaptation
associations were identified in the related network formed by the
miRNA, hsa-miR-21, and 2 related TFs, EGFR and nuclear factor of κ
light polypeptide gene enhancer in B-cells 1 (NFKB1). hsa-miR-21
also indirectly affects other miRNAs, including hsa-miR-141,
hsa-miR-106b and hsa-miR-222, by targeting the TFs that regulate
these miRNAs.
| Table II.Upstream and downstream genes of
hsa-miR-21 in testicular cancer. |
Table II.
Upstream and downstream genes of
hsa-miR-21 in testicular cancer.
| Location | Global network | Related network |
Differentially-expressed network |
|---|
| hsa-miR-21 |
|
|
|
|
Upstream | EGFR, ESR1, ETV5,
Foxo3a, AP-1, NFIB, NFKB1, PTEN, RAS/ERK, REL, RELA, REST, BMP6,
BMPR1a, BMPR1b, STAT3, TCF4 | MIR21, BMP6,
EGFR, ESR1, NFKB1, PTEN, REL,
RELA | MIR21,
PTEN |
|
Downstream | APAF1, BASP1, BCL2,
BMPR2, BTG2, CCR1, CDC25A, CDK2AP1, DAXX, DERL1, E2F1, E2F2, EGFR,
EIF2S1, EIF4A2, ERBB2, FMOD, HNRNPK, ICAM1, IL1B, ISCU, JAG1, JMY,
LRRFIP1, MARCKS, MEF2C, MSH2, MSH6, MTAP, MYC, NCAPG, NCOA3 PCBP1,
PDCD4, PDHA2, PLAT, PLOD3, PPIF, PTEN, PTX3, RASGRP1, RECK, REST,
RHOB, RPS7, RTN4, SERPINB5, SOX5, SPATS2L, SPRY2, TGFBI, TGFBR2,
TGFBR3, TGIF1, TIAM1, TIMP3, TM9SF3, TNFAIP3, TOPORS, TP53BP2,
TP63, TPM1, WFS1, WIBG | APAF1, E2F1,
E2F2, EGFR, MSH2, NFKB1, PPARA, PTEN | MSH2,
PTEN |
As a further example, it was identified that
hsa-miR-222 targets 2 TFs (PTEN and TP53), but is not regulated by
any TFs in the differentially-expressed network. Two
self-adaptation associations are derived in the regulated network
by importing related elements. hsa-miR-222 and hsa-miR-21 have all
three kinds of predecessors (upstream) and three types of
successors (downstream) adjacent nodes. Other miRNAs belonging to
this class include hsa-miR-96 and hsa-miR-141. These miRNAs are
considered as the first class of miRNAs, as they have 6 adjacent
nodes (3 successors and 3 predecessors) in the whole network.
Similarly to hsa-miR-302 and hsa-miR-145, other
classes of miRNAs also have 5 adjacent nodes (2 successors and 3
predecessors). These miRNAs are considered the second class of
miRNAs. The third class of miRNAs has 4 adjacent nodes (1 successor
and 3 predecessors), and includes hsa-miR-367, hsa-miR200c and
certain single miRNAs adjacent to the major nodes in the
differentially-expressed network, such as hsa-miR-187 and
hsa-miR-371. This information was derived through comparisons of
Figs. 1 and 2.
Regulatory pathways of popular
TFs
The P-match method was used to predict popular TFs
in TC, in addition to the identification of additional pathways
that may be involved in the process of differential expression.
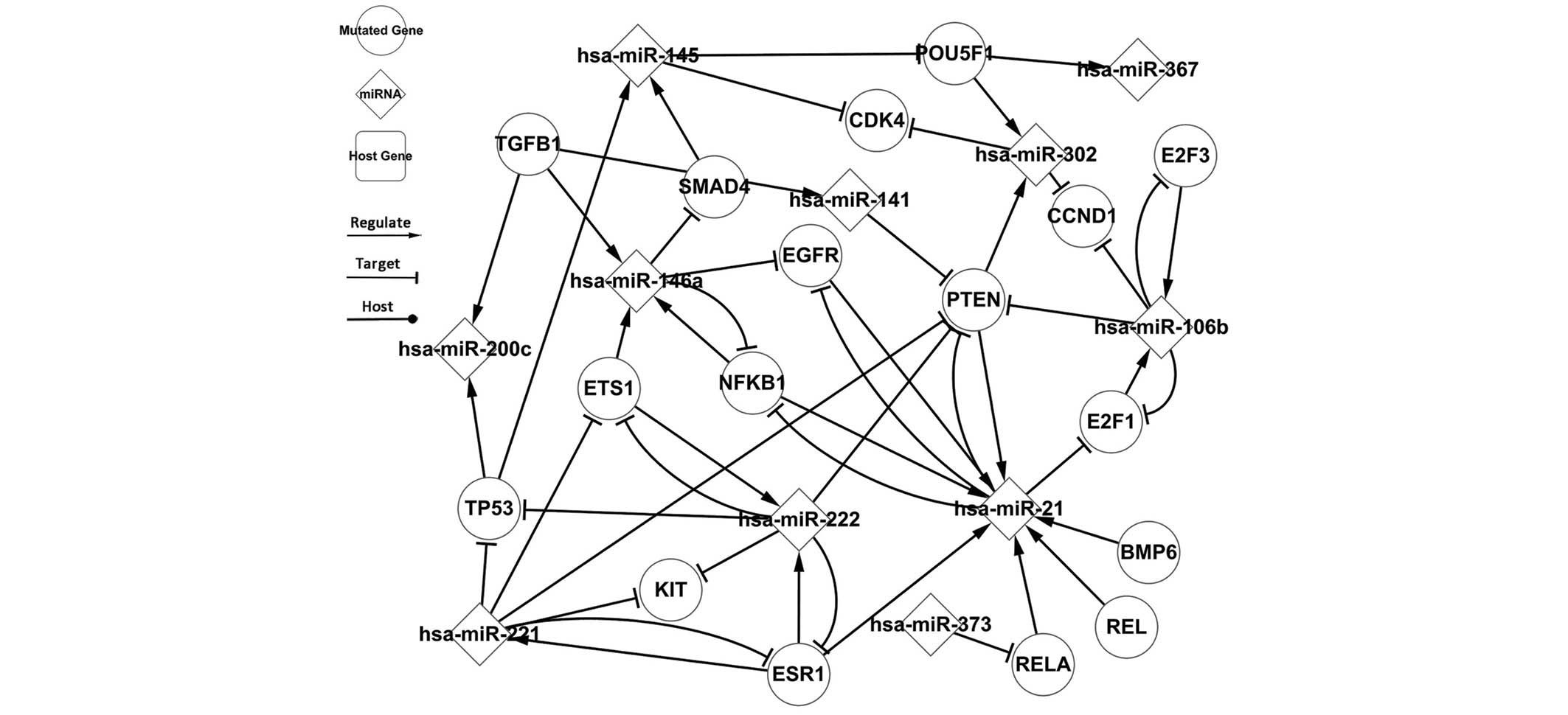
Fig. 3 presents the highlighted
regulatory pathways, composed of miRNAs and popular TFs, which were
derived from the related network. The related network primarily
underlines important regulatory relationships beyond the
differentially-expressed network. A total of 9 self-adaptation
associations are presented in the related network (Fig. 2), and involve the following TFs: PTEN,
E2F1, E2F3, EGFR, ETS1, ESR1 and NFKB1, demonstrating that these
TFs and miRNAs regulate one another.
The associations of NFKB1, a TF, were predicted
using the P-match method, for which the up- and downstream
information is presented in Table
III. NFKB1 is involved in 3 self-adaptation associations in the
related network, which includes hsa-miR-21, hsa-miR-146a and
hsa-miR-9. Firstly, hsa-miR-21 is differentially-expressed.
Secondly, whilst hsa-miR-146a is not differentially-expressed, it
targets a differentially-expressed TF (SMAD4) and was therefore
included as a self-adaptation association in the network. The
global network includes more than just information on TC, therefore
the global differences in the upstream and downstream elements can
be disregarded. The upstream and downstream elements of NFKB1 are
therefore the same regardless of the global network. This indicates
that NFKB1 is a self-adapting TF in the TC regulatory pathway.
| Table III.Upstream and downstream miRNAs of the
NFKB1 transcription factor in testicular cancer. |
Table III.
Upstream and downstream miRNAs of the
NFKB1 transcription factor in testicular cancer.
| Location | Global network | Related network |
Differentially-expressed network |
|---|
| NFKB1 |
|
|
|
|
Upstream | hsa-let-7a,
hsa-miR-9, hsa-miR-146a, hsa-miR-146a, hsa-miR-15a, hsa-miR-21,
hsa-miR-9, hsa-miR-146b-5p | hsa-miR-9,
hsa-miR-146a, hsa-miR-21 | hsa-miR-146a,
hsa-miR-21 |
|
Downstream | hsa-let-7a-3,
hsa-let-7b, hsa-miR-10b, hsa-miR-125b, hsa-miR-146a, hsa-miR-155,
hsa-miR-16, hsa-miR-17, hsa-miR-199a-2, hsa-miR-21, hsa-miR-214,
hsa-miR-224, hsa-miR-29a, hsa-miR-29b, hsa-miR-29c, hsa-miR-34a,
hsa-miR-365, hsa-miR-448, hsa-miR-9 | hsa-miR-9,
hsa-miR-146a, hsa-miR-21 | hsa-miR-146a,
hsa-miR-21 |
Certain TFs regulate other genetic elements but are
never targeted by miRNAs such as TGFB1, which regulates 3 miRNAs,
including 2 differentially-expressed miRNAs (hsa-miR-141 and
hsa-miR-200c). This indicates that TGFB1 may be located at the
bottom level of the regulatory pathway. As an additonal example,
POU5F1 is a gene that not differentially-expressed; however, all of
the adjacent, interacting nodes of POU5F1 are
differentially-expressed. Compared with the
differentially-expressed network, the regulatory pathway links
separated nodes together. The associated nodes further improve the
regulatory pathway and the combined action of each node shows the
regulatory progress and reveals pathology of TC.
Discussion
The present study derived three topological,
regulatory networks composed of elements associated with TC. The
key features of each pathway were discussed in detail, and
regulatory pathways were highlighted, which included important
elements involved in the regulatory process. Precursors and
successors were listed for the major elements identified in the
networks, and self-adaptation associations were identified.
Furthermore, any particular nodes that warranted further research
were indicated, and any predictions made using the P-match method
may provide additional aspects of research.
Differentially-expressed elements and the
associations between them are of particular importance, as it is
suspected that these elements may be the primary cause of TC. If
the relevant genes are differentially-expressed as described in the
network, then it is expected that TC may develop. If these elements
are able to be regulated back within normal ranges, then this may
prevent the occurrence of TC.
The current study identified a number of key
pathways involving differentially-expressed elements, which
included three or more of these elements. For example, TP53
associates with 3 differentially-expressed miRNAs, regulating two
of these (hsa-miR-200c and hsa-miR-145) and being targeted by one
(hsa-miR-222). These pathways are critical in TC pathogenesis and
illustrate a topological network of TC development. Certain
pathways have not only been identified in TC, but also in other
types of cancer. For example, the association whereby hsa-miR-21
targets PTEN also affects lung squamous cell carcinoma (18). Therefore, these associations between
genes may be extended from one form of cancer to another.
In conclusion, the present study used the P-match
method to predict TFs involved in TC pathogenesis, which suggested
that potential associations may exist between
differentially-expressed miRNAs and TFs. It is proposed that these
differentially-expressed elements may be manipulated to achieve
normal expression levels, subsequently preventing the development
of TC. However, these associations remain to be experimentally
validated in future studies, with the aim of eventually improving
the prognosis, diagnosis and treatment of TC.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 60973091 and
60905022) and the Natural Science Foundation of Jilin Province,
China (no. 201330101166JC).
Glossary
Abbreviations
Abbreviations:
|
miRNA
|
microRNA
|
|
TFs
|
transcription factors
|
|
NCBI
|
national center for biotechnology
information
|
|
TC
|
testicular cancer
|
References
|
1
|
American Cancer Society, . Testicular
cancer. http://www.cancer.org/cancer/testicularcancer/indexJanuary
15–2015
|
|
2
|
Eble JN, Sauter G, Epstein JI and
Sesterhenn IA: World Health Organization Classification of
TumoursPathology and Genetics of Tumours of the Urinary System and
Male Genital Organs. IARC Press; Lyon: 2004
|
|
3
|
Hayes-Lattin B and Nichols CR: Testicular
cancer: A prototypic tumor of young adults. Semin Oncol.
36:432–438. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hobert O: Gene regulation by transcription
factors and microRNAs. Science. 319:1785–1786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodriguez A, Griffiths-Jones S, Ashurst JL
and Bradley A: Identification of mammalian microRNA host genes and
transcription units. Genome Res 14 (10A). 1902–1910. 2004.
View Article : Google Scholar
|
|
6
|
Baskerville S and Bartel DP: Microarray
profiling of microRNAs reveals frequent coexpression with
neighboring miRNAs and host genes. RNA. 11:241–247. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cao G, Huang B, Liu Z, Zhang J, Xu H, Xia
W, Li J, Li S, Chen L, Ding H, et al: Intronic miR-301 feedback
regulates its host gene, ska2, in A549 cells by targeting MEOX2 to
affect ERK/CREB pathways. Biochem Biophys Res Commun. 396:978–982.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang J, Lu M, Qiu CU and Cui Q: TransmiR:
A transcription factor-microRNA regulation database. Nucleic Acids
Res. 38:(Database issue). D119–D122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kozomara A and Griffiths-Jones S: miRBase:
Integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:(Database issue). D152–D157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Safran M, Dalah I, Alexander J, Rosen N,
Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, et al:
GeneCards Version 3: The human gene integrator. Database (Oxford).
2010:baq0202010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vladušić T, Hrašćan R, Krušlin B,
Pećina-Šlaus N, Perica K, Bićanić A, Vrhovac I, Gamulin M and
Franekić J: Histological groups of human postpubertal testicular
germ cell tumours harbour different genetic alterations. Anticancer
Res. 34:4005–4012. 2014.PubMed/NCBI
|
|
12
|
Chekmenev DS, Haid C and Kel AE: P-Match:
Transcription factor binding site search by combining patterns and
weight matrices. Nucleic Acids Res. 33:(Web Server Issue).
W432–W437. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fujita PA, Rhead B, Zweig AS, Hinrichs AS,
Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A,
et al: The UCSC Genome Browser database: Update 2011. Nucleic Acids
Res. 39:(Database Issue). D876–D882. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang Q, Wang Y, Hao Y, Juan L, Teng M,
Zhang X, Li M, Wang G and Liu Y: miR2Disease: A manually curated
database for microRNA deregulation in human disease. Nucleic Acids
Res. 37:(Database Issue). D98–D104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Petitjean A, Achatz MIW, Borresen-Dale AL,
Hainaut P and Olivier M: TP53 mutations in human cancers:
Functional selection and impact on cancer prognosis and outcomes.
Oncogene. 26:2157–2165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bouras M, Tabone E, Bertholon J, Sommer P,
Bouvier R, Droz JP and Benahmed M: A novel SMAD4 gene mutation in
seminoma germ cell tumors. Cancer Res. 60:922–928. 2000.PubMed/NCBI
|
|
17
|
Di Vizio D, Cito L, Boccia A, Chieffi P,
Insabato L, Pettinato G, Motti ML, Schepis F, D'Amico W, Fabiani F,
et al: Loss of the tumor suppressor gene PTEN marks the transition
from intratubular germ cell neoplasias (ITGCN) to invasive germ
cell tumors. Oncogene. 24:1882–1894. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu LF, Wu ZP, Chen Y, Zhu QS, Hamidi S and
Navab R: MicroRNA-21 (miR-21) regulates cellular proliferation,
invasion, migration and apoptosis by targeting PTEN, RECK and Bcl-2
in lung squamous carcinoma, Gejiu City, China. PLoS One.
9:e1036982014. View Article : Google Scholar : PubMed/NCBI
|