Introduction
The wide application of next-generation sequencing
has identified millions of somatic alterations in cancer genomes
(1). Certain alterations responsible
for oncogenesis are termed driver mutations, but the majority
remain passenger mutations, which accumulate and have little
function in cancer progression (2).
At present, there are numerous bioinformatics tools available on
driver mutation prediction; the tools mostly focus on coding
mutations that change the amino acid residues, for example the
sorting tolerant from intolerant algorithm (3) and polymorphism phenotyping tool
(4). By contrast, there are few
studies conducted on the evaluation of the functional impact of
non-coding variants, and identification of non-coding drivers in a
typical tumor is a challenging and unsolved problem.
Recently, the interpretation of non-coding variants
has been achievable due to the production of high-throughput
projects, such as the Encyclopedia of DNA Elements (ENCODE)
Consortium (5) and the US National
Institutes of Health Roadmap Epigenomics project (6). Based on these data, a number of tools
have been developed to annotate potential regulatory variants or
suggest the most likely damaging variants, such as RegulomeDB
(7), HaploReg (8) and Funseq (9). Despite the high efficiency of functional
annotation of non-coding variants with these tools, there have been
certain criticisms of the empirical scoring algorithms, such as
lack of accuracy and specificity (10). Recently, machine-learning models were
introduced and trained on pathogenic variants or nearly-fixed/fixed
human derived alleles to better predict and score the
functionalities of non-coding variants (11,12). Fu
et al (13) reported a
computational framework, FunSeq2, that combines an adjustable data
context integrating large-scale genomics, such as 1000 Genomes and
ENCODE data, and cancer resources with a weighted scoring system.
Variants are scored by combining inter– and intra-species
conservation, loss– and gain-of-function events for
transcription-factor binding, enhancer-gene linkages and network
centrality, and per-element recurrence across samples (13). Kircher et al (11) contrasted the annotations of fixed or
nearly-fixed derived alleles in humans with those of simulated
variants and developed combined annotation-dependent depletion
(CADD). CADD, as a trained support vector machine, measures
deleteriousness, which may be measured systematically across the
genome assembly (14). Implementation
of CAAD successfully differentiated 14.7 million high-frequency
human-derived alleles from 14.7 million simulated variants.
Long non-coding RNAs (lncRNAs) are a class of
mRNA-like transcripts ranging between 200 bp and 100 kb in size.
lncRNAs lack significant open-reading frames and are not translated
into proteins. There has been a large number of studies that
reported lncRNAs to be involved in a wide range of physiological
processes by regulating gene expression at various levels,
including chromatin architecture, transcription, RNA splicing, and
protein translation and turnover (15–19).
Previously, the role of lncRNAs as drivers of tumor suppressive and
oncogenic functions has been reported in prevalent cancer types.
For example, HOX transcript antisense RNA (HOTAIR) expression is
high in breast cancer tumors that are predisposed to metastasize,
and the inhibition of HOTAIR expression blocks metastasis in mouse
models (17). Metastasis associated
lung adenocarcinoma transcript 1 (MALAT1) expression correlates
with metastasis and overall survival rate in lung cancer (18). An increasing number of studies have
explored methods to identify non-coding driver genes in cancers
(19–21). Du et al (19) selected lncRNAs in recurrent somatic
copy-number alterations (amplification) regions as candidate
drivers. Knockdown of either prostate cancer-associated non coding
RNA 1 (PCAN-R1) or 2 (PCAN-R2), which are the two most notably
differentially expressed lncRNAs between normal prostate tissue and
primary prostate cancer, resulted in substantial decreases in cell
growth and colony formation in the androgen-dependent prostate
cancer LNCaP cell line, suggesting that PCAN-R1 and PCAN-R2 have
tumor-promoting functions in prostate cancer (19).
While numerous studies have investigated generalized
variants (7,9,11,12), this is not the case for
cancer-specific somatic mutations. In the present study, a liver
cancer-specific annotation, mainly from the ENCODE project, was
used to investigate whether a combination of non-coding features
may be predictive for non-coding pathogenic variants. This scoring
system was then added to prioritize cancer-associated lncRNAs in
liver cancer.
In a previous study, we successfully constructed a
logistic regression model to score the functionalities of
non-coding variants using an array of lung cancer-specific features
(22). Considering the function
impact of non-coding variants is largely feature and cancer
type-dependent, in the present study, another logistic regression
model was created based on a liver cancer-specific feature
annotation to interpret the function information of non-coding
pathogenic variants. Subsequently, this scoring system was applied
to prioritize cancer-associated lncRNAs in liver cancer.
Materials and methods
Cancer mutation and pathogenic variant
data
A total of 881,136 somatic mutations of human liver
cancer were detected by whole-genome sequencing of 88 pairs of
cancer and normal tissues. This data was obtained from the
supplementary data files of the study by Alexandrov et al
(23). Recurrent cancer mutation was
defined as mutations that are recurrently mutated at least two
times at the same site across multiple samples. Non-recurrent
mutation denotes mutations that only occur once in all patients. In
total, 1,121 mutations of liver cancer were defined as recurrent
and the remaining mutations were considered to be non-recurrent.
Germline polymorphism data comprising 38,248,779 single nucleotide
polymorphisms (SNPs) was downloaded from the 1000 Genome project
pilot 1 (24). SNPs with a derived
allele frequency ≤0.01 were considered neutral SNPs. Rare SNPs are
the SNPs with an allele frequency <0.01. Disease-associated
variants data contained in ClinVar and the human gene mutation
database (HGMD) are published gene variants responsible for human
inherited diseases (25,26). Genome-wide association study (GWAS)
SNPs from GWAS (27) are numerous
common genetic variants associated with a trait or disease.
Genome-wide feature sets
Human genome annotations were obtained from Gencode
(28), including protein coding
genes, exons, introns, lncRNAs, lncRNA exons, introns, untranslated
regions (UTRs) and non-coding exons (ncExon) (28). The 5′ splicing sites are 10
nucleotides from the 5′ end of the introns of genes. The 3′
splicing sites are 50 nucleotides from the 3′ end of the introns of
genes (29). Evolutionarily conserved
bases were identified using a recently published analysis of 46
mammalian genomes (30). Genome-wide
phastCons scores were obtained from the study by Siepel et
al (31). Sensitive regions from
the study by Khurana et al (9)
consist of binding sites or motifs of important transcription
factors and contain a higher fraction of rare SNPs. Evolutionarily
conserved structures (ECSs) from the study by Smith et al
(32) are RNA secondary structures
predicted using comparative structure prediction algorithms based
on multiple genomes. Promoters, which are regions 2.5 kb from the
transcription start sites, were generated by the Gerstein lab and
are publicly available for download (9). RNA sequencing (RNA-seq) data in bam
format, transcription factor binding sites (TFBSs), DNase I
hypersensitive sites (DNase I), histone modification data,
including H3K4me1 and H3K9ac, of the Hepg2 cell line were acquired
from ENCODE (33). Conserved TFBSs
are transcription factor binding sites conserved in the
human/mouse/rat alignment and obtained from UCSC directly (30). The expression level was calculated by
counting the number of reads per kb per million reads (RPKM) for
each protein coding gene and lncRNA. Genes with a RPKM >20 or
<0.25 were defined as high and low-expressed regions,
respectively. A wavelet-smoothed, weighted average signal, with
high and low values that indicate early and late replication during
the S phase, respectively (33), was
used in the present study. Genome-wide replication timing was
mapped to protein coding genes and lncRNAs, and an early-to-late
ratio was calculated for each protein coding gene and lncRNA, as
follows: Early-to-late ratio = (G1b + S1) / (S4 + G2). As the
early-to-late ratio is >1, genes are considered early
replicated, while late replicated genes have an early-to-late ratio
<1.
Cancer lncRNAs, consisting of 25 lncRNAs, are a
curation of mammalian long non-coding transcripts that have been
experimentally shown to be associated with a variety of cancer
types (22). A list of cancer census
genes was obtained from the current release of catalogue of somatic
mutations in cancer (COSMIC) v71 (34).
Logistic regression model
The disease-implicated set of variants was composed
of all pathogenic variants from the ClinVar and HGMD databases.
Subsequent to the removal of coding variants, a set of 19,153
non-coding disease-implicated single nucleotide variants (SNVs)
remained. For the control sets, neutral variants with a minor
allele frequency ≥1% were used to reduce the possibility of
including functional rare SNPs. A total of 15,789,242 potential
control SNVs were included in the model. In the logistic regression
model, a matrix of 395,279 rows was formed throughout the
non-coding genome; each row represents one type of combination of
features. A disease-implicated set of variants was used as success
(disease-causing variant), neutral SNPs were used as control, and
the 26 genomic binary features were used as the predictor variable.
The logistic regression model was constructed using a general
linear model, and the receiver operating characteristic (ROC) curve
was generated using a script in R generated by the present authors.
The scores were predicted using the model for GWAS, neutral SNPs,
non-recurrent and recurrent somatic mutations of liver cancer and
then scaled using the following formula: Scaled score = log
(predicted score × 106).
Prioritization of cancer-associated
lncRNA candidates
In order to filter out potential functional lncRNAs
involved in liver cancer, two different strategies were utilized.
First, the fraction of high scoring regions and the average score
for each lncRNA were calculated. Secondly, the top 10% of lncRNAs,
which contained the highest fraction of high-scoring regions, and
the 10% lncRNAs that had the highest average score were determined,
and a subset of overlapping lncRNAs were generated by intersection
of the two different lncRNAs sets, forming 847 functional lncRNA
candidates, and the remaining lncRNAs were considered to be control
lncRNAs.
RNA-seq data processing and expression
analyses
The RNA-seq data of 12 pairs of liver cancer samples
were obtained from the study by Zhang et al (GSE63863)
(35). The reads were aligned to the
hg19 genome using TopHat2 version 2.0.13 (36). Read counts were calculated with
BEDTools v2.22.1 for each lncRNA (37). The relative expression level was
calculated as the RPKM + 1 and then log scaled for each lncRNA.
DESeq2 Release (3.0) (38) was used
to identify differentially expressed transcripts between tumor and
normal pairs, with a false discovery rate (FDR) cutoff of ≤10-4 and
absolute fold change cutoff of ≥1.5.
Statistical analyses
The data were expressed as the mean values. The
difference between groups was tested using the two-sided
Mann-Whitney rank sum test (wilcox.test) or Fisher's exact test
(fisher.test) in R. P<0.05 was considered to indicate a
statistically significant difference.
Results
Logistic regression model successfully
discriminates between functional non-coding variants and neutral
variants
Density of disease-causing variants estimates showed
that different features exhibit differential enrichment of
deleterious variants (Fig. 1A).
Conserved regions, conserved TFBS, UTRs, high-expressed regions and
promoters showed the highest densities of disease-causing variants.
By contrast, H3K9me3, late-replicated regions, ECSs, H2az and
H3K27me3 are the least enriched features in disease-causing
variants, indicating that various non-coding features have varied
importance to the functionalities of non-coding variants. The
present study used a logistic regression model to build a
classifier to discriminate between the disease-associated and
control variants. The present study analyzed the features that
contribute most to the discriminative power of the present model
(Fig. 1B). Generally, the present
study observed that conserved regions, early-replicated regions,
promoters, H3K36me3 and conserved TFBSs are the most positive
factors contributing to the model, while H3K9me3, H3K79me2,
H4K20me1 and ncExon are the most negative factors affecting the
prediction capability of the model. The ROC curve for the
classifier is shown in Fig. 1C. The
area under the ROC curve (AUC) is 0.92, which demonstrates that the
present model may discriminate between disease-implicated and
control variants with a high specificity and sensitivity.
 | Figure 1.(A) Density of disease-causing
variants from the ClinVar database and human gene mutation database
associated with different genome features (red line, the average in
the human genome). Different features exhibit differential
enrichment of deleterious variants, with conserved regions highest
and H3K9me3 lowest. (B) Regression coefficient for each feature.
(C) Receiver operating characteristic curve for the logistic
regression model. (D) Predicted scores for GWAS, neutral SNPs, and
non-recurrent and recurrent somatic mutations of liver cancer. The
scores were scaled using the formula ‘scaled score = log (predicted
score ×106)’. GWAS disease associated variants and recurrent
somatic mutations of liver cancer showed elevated average scores as
compared with neutral SNPs and non-recurrent somatic mutations
respectively. TFBS, transcription factor binding site; cTFBS,
conserved TFBS; UTR, untranslated region; CR, conserved region;
SNP, single nucleotide polymorphism; Sensitive, known regions with
a high ratio of rare SNP (allele frequency <0.01); ER, early
replicated regions; LR, late replicated regions; HE, high expressed
regions; LE, low expressed regions; ECS, evolutionarily conserved
structure; Dnase I, Dnase I hypersensitive site; H3K/H4K, histone
modification data; ncExon, non-coding exon; TPR, true positive
rate; FPR, false positive rate. |
To establish whether the present prediction scores
are likely to be generalizable to other data sets, the current
study conducted experiments that demonstrate how the predicted
scores may be applied to prioritize candidate functional variants.
For the first experiment, non-coding variants associated with
complex disease from genome-wide association studies (GWAS) were
annotated. It was found that non-coding GWAS SNVs had a
significantly higher average score compared with control variants
(mean score, 5.8642 vs. 5.4707; P<0.001; two-sided Mann-Whitney
U test; Fig. 1D).
Recurrence is considered to be one potential sign of
positive selection among tumors and is more likely to be associated
with driver events (33). As an
application to cancer studies, the present study annotated
non-coding somatic mutations identified in whole-genome sequencing
studies from 88 liver cancer samples. The present study identified
recurrent somatic mutations that had occurred at the same site in
multiple samples (n=1,121 mutations) and found that these recurrent
mutations were assigned a significantly higher average score
compared with non-recurrent mutations (5.4101 vs. 5.2768; P=0.0125,
Mann-Whitney U test; Fig. 1D). This
finding demonstrates that this approach may be useful in the
detection of cancer driver mutations in liver cancer.
High-scoring regions define cancer
‘hotspots’ in introns, UTRs and lncRNAs
The present study defined 100-mb non-coding regions
that had the highest scores predicted by the model as high-scoring
regions, and gene features with the highest fractions of
high-scoring regions were sought. Features with the highest
fractions of high scoring regions included intronic 5′ and 3′
splice sites and 3′-UTRs (Fig. 2A).
Splicing sites were scored significantly higher compared with
adjacent intronic regions in protein coding genes (9.3463 vs.
8.3263; P<0.01; Fig. 2B) and
lncRNAs (8.2544 vs. 7.9248; P<0.001; Fig. 2B). Furthermore, known cancer genes
from COSMIC have significantly more high-scoring regions in the
gene introns and UTRs compared with non-cancer genes (0.0850 vs.
0.0640; P<0.001; Fig. 2C). In
general, lncRNAs do not contain a large fraction of high-scoring
regions (Fig. 2C). However, the known
cancer-associated lncRNAs showed a significantly increased fraction
of high-scoring regions as compared to general lncRNAs (0.0997 vs.
0.0404; P<0.001; Fig. 2C). For
example, HOTAIR and MALAT1 are among the top 10% of lncRNAs with
respect to overlap with high-scoring regions (Fig. 2D).
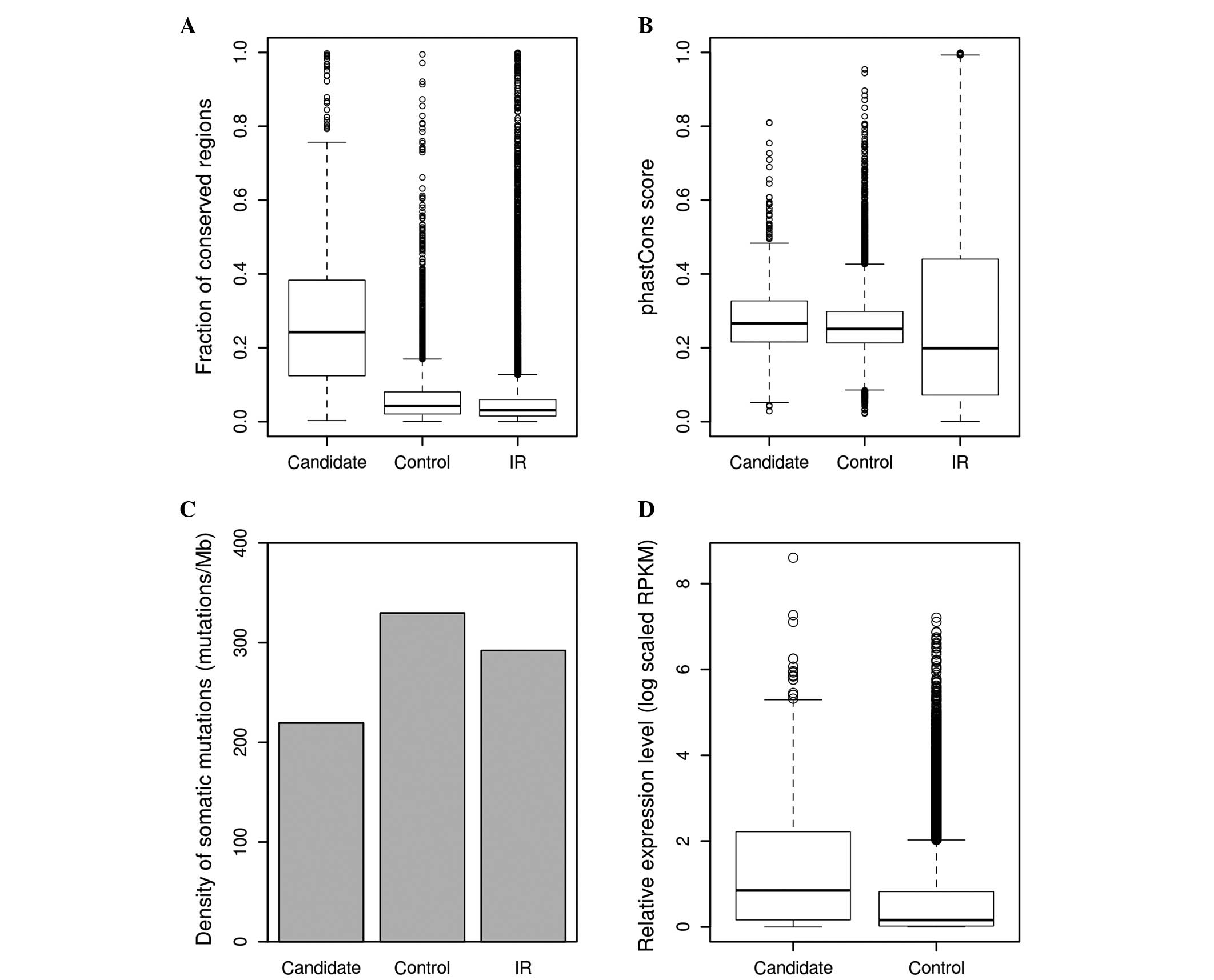
Prioritization of liver
cancer-associated lncRNAs with the scoring system
In the present study, 847 lncRNAs were identified by
combining the fraction of high-scoring regions and the average
score of lncRNAs. Among these are lncRNAs that are known to be
cancer-associated, such as MALAT1, HOTAIR and growth arrest
specific 5 (GAS5). This final subset of lncRNAs was found to be
significantly more conserved compared with other control lncRNAs,
in terms of fraction of conserved regions (0.1635 vs. 0.0536;
P<0.001; Fig. 3A) and phastCons
score (0.2813 vs. 0.2644; P<0.001; Fig. 3B). It was also observed that this
subset of lncRNAs had a lower somatic mutation density compared
with the control lncRNAs (219.4753 vs. 329.7922 mutations/Mb;
P<0.001; Fig. 3C); RNA-seq data of
12 pairs of liver cancer samples were obtained from the study by
Zhang et al (35). Read
alignment was conducted using TopHat2 and coverage was calculated
for each lncRNAs using BEDTools. It was found that the list of
lncRNA candidates had increased average expression levels compared
with the control lncRNAs in cancer and normal samples [log scaled
(RPKM+1), 1.3868 vs. 0.6327; P<0.001; Fig. 3D]; DESeq2 was used to evaluate the
different expression of lncRNAs in 12 pairs of liver cancer and
adjacent normal samples. lncRNAs with FDR ≤10-4 and absolute fold
change ≥1.5 were considered to be differentially expressed. In
total, there were 353 lncRNAs that met the selection criteria, 23
of which are among the list of potentially cancer-associated
lncRNAs (Fig. 4).
Discussion
It is evident that only a small subset of genetic
variations contributes to tumor evolution by providing cells with a
selective advantage over their neighbors (10). The damaging impact of coding mutations
may be evaluated efficiently with a variety of tools; however,
functional annotation of mutations in the non-coding fraction of
the human genome is markedly more obscure and challenging.
Recently, GWAVA used all variations annotated as
‘regulatory mutations’ from the public release of HGMD and combined
annotations to build three random forest classifiers that
prioritize disease-associated variants (12); however, this study focused on
regulatory mutations of the HGMD database and predicted regulatory
pathogenic variants, which was incomplete and limited in scope to
the regulatory regions. The present logistic regression model
included all non-coding pathogenic variants from HGMD and ClinVar
databases, which allows for evaluation of damaging impact of any
variant in the non-coding genome. Furthermore, a liver
cancer-specific annotation was used, which facilitated the
identification of driver mutations in a liver cancer-specific
fashion. It was found that non-coding features, such as conserved
regions, early replicated regions, promoter, H3K36me3, conserved
TFBS, sensitive regions, TFBS, H3K4me3 and H3K9ac are among the
positive factors that most contribute to the model. Histone
modifications, such as H3K4me3 and H3K9ac, are hallmarks of
actively transcribed protein-coding promoters in eukaryotes, and
H3K36me3 has long been associated with the gene bodies of actively
transcribed genes (40). All these
findings support the hypothesis that conserved regions and
regulatory elements play notable roles in the formation and
functionality of pathogenic variants in the non-coding genome.
The AUC demonstrates how well a classifier can
discriminate between disease and control variants; the AUC of the
present logistic regression model was as high as 0.92, which showed
a reliable and high-efficient performance. Furthermore, the present
study showed the utility of the present model by providing two
types of examples of common experiments using disease or
trait-associated GWAS variants and recurrent cancer mutations. The
present study demonstrates that the present model effectively
discriminates non-coding GWAS SNVs from control variants.
Recurrence of somatic mutations is a widely used proxy of likely
function; the present scoring system scored recurrent mutations
significantly higher compared with non-recurrent mutations,
suggesting this approach may allow for the identification of cancer
driver mutations.
With respect to the distribution of high scoring
regions identified by the present model, it was found that splicing
sites of either protein coding genes or lncRNAs and UTRs were most
enriched with the highest fraction of high scoring regions, as
these regions are highly evolutionarily conserved across mammals
(41). Notably, it was found that
known cancer genes and cancer-implicated lncRNAs contain a higher
fraction of high-scoring regions compared with
non-cancer-associated counterparts. Two typical examples are MALAT1
and HOTAIR, which have been involved in the tumorigenesis and
progression in a variety of cancer types (17,18,42–47).
The fraction of high scoring regions and average score predicted by
the present model was combined for each lncRNA and filtered out the
a subset of functional lncRNA candidates, which include
experimentally characterized functional lncRNAs, such as MALAT1,
HOTAIR, HOXA transcript antisense RNA, myeloid-specific 1 and GAS5.
The present study found that this small subset of lncRNAs are more
conserved, less mutated and demonstrate increased expression
compared with the control lncRNAs, and 23 of the 847 lncRNAs
identified are differentially expressed in 12 pairs of liver cancer
and normal samples. These lncRNAs are notable candidates for
experimental validation and characterization in future studies.
Overall, the present study defined a scoring system
for evaluating the damaging effect of non-coding variants in liver
cancer. This system allows the identification of putative harmful
mutations in a liver-cancer specific fashion in the introns and
UTRs of mRNAs, as well as prioritizing a number of lncRNA
candidates for additional experimental validation.
Acknowledgements
The present study was supported by the National
Natural Sciences Foundation of China (grant no, 81272142; to Xin
LV).
References
|
1
|
Robison K: Application of
second-generation sequencing to cancer genomics. Brief Bioinform.
11:524–534. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Greenman C, Stephens P, Smith R, Dalgliesh
GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C,
et al: Patterns of somatic mutation in human cancer genomes.
Nature. 446:153–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sim NL, Kumar P, Hu J, Henikoff S,
Schneider G and Ng PC: SIFT web server: Predicting effects of amino
acid substitutions on proteins. Nucleic Acids Res. 40:(Web Server
Issue). W452–W457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
ENCODE Project Consortium, . An integrated
encyclopedia of DNA elements in the human genome. Nature.
489:57–74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bernstein BE, Stamatoyannopoulos JA,
Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra
MA, Beaudet AL, Ecker JR, et al: The NIH roadmap epigenomics
mapping consortium. Nat Biotechnol. 28:1045–1048. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boyle AP, Hong EL, Hariharan M, Cheng Y,
Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, et
al: Annotation of functional variation in personal genomes using
RegulomeDB. Genome Res. 22:1790–1797. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ward LD and Kellis M: HaploReg: A resource
for exploring chromatin states, conservation and regulatory motif
alterations within sets of genetically linked variants. Nucleic
Acids Res. 40:(Database Issue). D930–D934. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khurana E, Fu Y, Colonna V, Mu XJ, Kang
HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, et
al: Integrative annotation of variants from 1092 humans:
Application to cancer genomics. Science. 342:12355872013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Drubay D, Michiels S and Gautheret
D: Mining the coding and non-coding genome for cancer drivers.
Cancer Lett. 369:307–315. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kircher M, Witten DM, Jain P, O'Roak BJ,
Cooper GM and Shendure J: A general framework for estimating the
relative pathogenicity of human genetic variants. Nat Genet.
46:310–315. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ritchie GR, Dunham I, Zeggini E and Flicek
P: Functional annotation of noncoding sequence variants. Nat
Methods. 11:294–296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip
KY, Khurana E and Gerstein M: FunSeq2: A framework for prioritizing
noncoding regulatory variants in cancer. Genome Biol. 15:4802014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cooper GM, Stone EA and Asimenos G: NISC
Comparative Sequencing Program, Green ED, Batzoglou S and Sidow A:
Distribution and intensity of constraint in mammalian genomic
sequence. Genome Res. 15:901–913. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nie L, Wu HJ, Hsu JM, Chang SS, Labaff AM,
Li CW, Wang Y, Hsu JL and Hung MC: Long non-coding RNAs: Versatile
master regulators of gene expression and crucial players in cancer.
Am J Transl Res. 4:127–150. 2012.PubMed/NCBI
|
|
16
|
Gutschner T and Diederichs S: The
hallmarks of cancer: A long non-coding RNA point of view. RNA Biol.
9:703–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote cancer
metastasis. Nature. 464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ji P, Diederichs S, Wang W, Böing S,
Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et
al: MALAT-1, a novel noncoding RNA and thymosin beta4 predict
metastasis and survival in early-stage non-small cell lung cancer.
Oncogene. 22:8031–8041. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Du Z, Fei T, Verhaak RG, Su Z, Zhang Y,
Brown M, Chen Y and Liu XS: Integrative genomic analyses reveal
clinically relevant long noncoding RNAs in human cancer. Nat Struct
Mol Biol. 20:908–913. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mularoni L, Sabarinathan R, Deu-Pons J,
Gonzalez-Perez A and López-Bigas N: OncodriveFML: a general
framework to identify coding and non-coding regions with cancer
driver mutations. Genome Biol. 17:1282016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yan X, Hu Z, Feng Y, Hu X, Yuan J, Zhao
SD, Zhang Y, Yang L, Shan W, He Q, et al: Comprehensive Genomic
Characterization of Long Non-coding RNAs across Human Cancers.
Cancer Cell. 28:529–540. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H and Lv X: Functional annotation of
noncoding variants and prioritization of cancer-associated lncRNAs
in lung cancer. Oncol Lett. 12:222–230. 2016.PubMed/NCBI
|
|
23
|
Alexandrov LB, Nik-Zainal S, Wedge DC,
Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A,
Børresen-Dale AL, et al: Signatures of mutational processes in
human cancer. Nature. 500:415–421. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
1000 Genomes Project Consortium. Abecasis
GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang
HM, Marth GT and McVean GA: An integrated map of genetic variation
from 1,092 human genomes. Nature. 491:56–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Landrum MJ, Lee JM, Riley GR, Jang W,
Rubinstein WS, Church DM and Maglott DR: ClinVar: Public archive of
relationships among sequence variation and human phenotype. Nucleic
Acids Res. 42:(Database Issue). D980–D985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stenson PD, Mort M, Ball EV, Howells K,
Phillips AD, Thomas NS and Cooper DN: The human gene mutation
database: 2008 update. Genome Med. 1:132009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Beck T, Hastings RK, Gollapudi S, Free RC
and Brookes AJ: GWAS Central: A comprehensive resource for the
comparison and interrogation of genome-wide association studies.
Eur J Hum Genet. 22:949–952. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Harrow J, Frankish A, Gonzalez JM,
Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa
A, Searle S, et al: GENCODE: The reference human genome annotation
for the ENCODE project. Genome Res. 22:1760–1774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ward AJ and Cooper TA: The pathobiology of
splicing. J Pathol. 220:152–163. 2010.PubMed/NCBI
|
|
30
|
Karolchik D, Barber GP, Casper J, Clawson
H, Cline MS, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L,
Haeussler M, et al: The UCSC Genome Browser database: 2014 update.
Nucleic Acids Res. 42:(Database Issue). D764–D770. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Siepel A, Bejerano G, Pedersen JS,
Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW,
Richards S, et al: Evolutionarily conserved elements in vertebrate,
insect, worm and yeast genomes. Genome Res. 15:1034–1050. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smith MA, Gesell T, Stadler PF and Mattick
JS: Widespread purifying selection on RNA structure in mammals.
Nucleic Acids Res. 41:8220–8236. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rosenbloom KR, Sloan CA, Malladi VS,
Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R,
Heitner SG, et al: ENCODE data in the UCSC genome browser: Year 5
update. Nucleic Acids Res. 41:(Database Issue). D56–D63. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Forbes SA, Bindal N, Bamford S, Cole C,
Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al:
COSMIC: Mining complete cancer genomes in the catalogue of somatic
mutations in cancer. Nucleic Acids Res. 39:(Database Issue).
D945–D950. 011. View Article : Google Scholar
|
|
35
|
Zhang H, Weng X, Ye J, He L, Zhou D and
Liu Y: Promoter hypermethylation of TERT is associated with
hepatocellular carcinoma in the Han Chinese population. Clin Res
Hepatol Gastroenterol. 39:600–609. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim D, Pertea G, Trapnell C, Pimentel H,
Kelley R and Salzberg SL: TopHat2: Accurate alignment of
transcriptomes in the presence of insertions, deletions and gene
fusions. Genome Biol. 14:R362013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Quinlan AR and Hall IM: BEDTools: A
flexible suite of utilities for comparing genomic features.
Bioinformatics. 26:841–842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dees ND, Zhang Q, Kandoth C, Wendl MC,
Schierding W, Koboldt DC, Mooney TB, Callaway MB, Dooling D, Mardis
ER, et al: MuSiC: Identifying mutational significance in cancer
genomes. Genome Res. 22:1589–1598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hon GC, Hawkins RD and Ren B: Predictive
chromatin signatures in the mammalian genome. Hum Mol Genet.
18:R195–R201. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Washietl S, Kellis M and Garber M:
Evolutionary dynamics and tissue specificity of human long
noncoding RNAs in six mammals. Genome Res. 24:616–628. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang MH, Hu ZY, Xu C, Xie LY, Wang XY,
Chen SY and Li ZG: MALAT1 promotes colorectal cancer cell
proliferation/migration/invasion via PRKA kinase anchor protein 9.
Biochim Biophys Acta. 1852:166–174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shen L, Chen L, Wang Y, Jiang X, Xia H and
Zhuang Z: Long noncoding RNA MALAT1 promotes brain metastasis by
inducing epithelial-mesenchymal transition in lung cancer. J
Neurooncol. 121:101–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Okugawa Y, Toiyama Y, Hur K, Toden S,
Saigusa S, Tanaka K, Inoue Y, Mohri Y, Kusunoki M, Boland CR and
Goel A: Metastasis-associated long non-coding RNA drives gastric
cancer development and promotes peritoneal metastasis.
Carcinogenesis. 35:2731–2739. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Han Y, Liu Y, Zhang H, Wang T, Diao R,
Jiang Z, Gui Y and Cai Z: Hsa-miR-125b suppresses bladder cancer
development by down-regulating oncogene SIRT7 and oncogenic long
non-coding RNA MALAT1. FEBS Lett. 587:3875–3882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Deng Q, Sun H, He B, Pan Y, Gao T, Chen J,
Ying H, Liu X, Wang F, Xu Y and Wang S: Prognostic value of long
non-coding RNA HOTAIR in various cancers. PLoS One. 9:e1100592014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Endo H, Shiroki T, Nakagawa T, Yokoyama M,
Tamai K, Yamanami H, Fujiya T, Sato I, Yamaguchi K, Tanaka N, et
al: Enhanced expression of long non-coding RNA HOTAIR is associated
with the development of gastric cancer. PLoS One. 8:e770702013.
View Article : Google Scholar : PubMed/NCBI
|