Introduction
Angiogenesis refers to the formation of new
capillaries from pre-existing blood vessels (1). Solid tumor growth depends on
angiogenesis to supply nutrients and dispose of catabolic products
(2). Without new blood vessels, tumor
cells cannot sustain proliferation and thus are likely to remain
dormant (3). Anti-angiogenesis has
become a significant adjuvant treatment strategy in cancer
chemotherapy (3). Over the previous
decade, numerous anti-angiogenic agents have been developed, and
some of these have been approved by the FDA. These agents include
bevacizumab, sorafenib and sunitinib (4,5). However,
not all cancer patients benefit from existing anti-angiogenic
therapies (4–6). Novel anti-angiogenic strategies are
required to improve the prognosis of such patients.
Tumor angiogenesis is a tightly controlled process,
which is initiated by the secretion of growth factors, including
vascular endothelial growth factor (VEGF), from tumor cells
(7). These growth factors bind to
their receptors on endothelial cells and activate intracellular
signal transduction pathways to modify endothelial cell
proliferation, migration, apoptosis and differentiation (7). The mitogen-activated protein kinase
(MAPK) signaling pathway is a signaling cascade that mediates
diverse extracellular stimuli and regulates key cellular functions
(8). In mammals, MAPKs have three
major subfamilies: Extracellular signal-regulated kinase (ERK1/2 or
p44/42 MAPK), p38 MAPK and c-Jun N-terminal kinase
(JNK)/stress-activated protein kinase (SAPK) (9). JNK has three isoforms (JNK1, 2 and 3).
JNK1 and JNK2 are expressed in the majority of cell types,
including endothelial cells, while JNK3 is mainly expressed in
neuronal tissues (10–13). The JNK signaling pathway is activated
primarily by cytokines or exposure to environmental stresses
(10,14). These extracellular stimuli trigger the
activation of MAP kinase kinase kinases, which subsequently
phosphorylate the mitogen-activated protein kinase kinase isoforms
mitogen-activated protein kinase kinase (MKK)4 and MKK7. MKKs are
dual-specificity protein kinases that phosphorylate JNK at Thr183
and Tyr185 (10,14). JNK specifically phosphorylates the
transcription factor c-Jun on its N-terminal transactivation domain
at two serine residues, Ser63 and Ser73 (15). JNK and c-Jun activate transcription
factors, including activator protein 1, activating transcription
factor 2, Elk-1, p53 and c-Myc (16),
which consequently regulate downstream genes involved in apoptosis,
proliferation and differentiation (17,18). In
addition, the JNK cascade is modulated by Notch, nuclear factor
(NF)-κB and other signaling pathways (16,19).
Artemisinin is a lactonic sesquiterpenoid compound
originally isolated from Artemisia annua L and has been used
to treat malaria since the 1970s (20). An artemisinin derivative,
dihydroartemisinin (DHA), is more water-soluble and currently
considered the most effective drug in treating cerebral malaria
(21,22). DHA has also demonstrated strong
anti-tumor activity (23). Recently,
DHA emerged as a promising agent with potent anti-angiogenic
properties (24). DHA represses the
expression of VEGF in several cancer cell lines (23,25). In
cultured endothelial cells, DHA inhibits proliferation, migration
and tube formation (26–28). In a mouse model of retinal
neovascularization, intravitreal injection of DHA reduced
angiogenesis (24). As a widely used
anti-malarial drug, DHA has been proven to be safe with minimal
side effects and may be used clinically as a component of cancer
chemotherapy (29). However,
mechanistic studies of its anti-angiogenic effects at low
concentrations are limited. Previously, the present authors
reported that 20 µM of DHA (estimated C-max of the treatment of 12
mg/kg body weight recommended by the World Health Organisation for
antimalarial therapy) inhibits endothelial cell proliferation by
suppression of phosphorylation and protein expression of ERK1/2
(30), whereas it does not affect p38
MAPK in endothelial cells (31). The
role of the JNK signaling pathway in the response of endothelial
cells to DHA remains unclear.
In the present study, the effects of DHA on JNK
signaling were investigated in human umbilical vein endothelial
cells (HUVECs). At a concentration of 20 µM, DHA transiently
activated JNK and increased the expression of JNK signaling pathway
downstream genes. An identical concentration of DHA inhibited the
NF-κB signaling pathway, but did not affect endothelial cell
apoptosis. The results of the present study may improve our
understanding of the molecular mechanisms of the anti-angiogenic
activities of DHA.
Materials and methods
Cell culture
HUVECs were purchased from Lonza (Basel,
Switzerland) and cultured in basal endothelial cell medium (EBM2)
supplemented with EGM-2-MV bullet kit (Lonza) and antibiotics (100
IU/ml penicillin and 100 µg/ml streptomycin). The cells were placed
in humidified air at 37°C with 5% CO2. DHA (D7439;
Sigma-Aldrich; EMD Millipore, Billerica, MA, USA), pyrrolidine
dithiocarbamate (PDTC; P8765; Sigma-Aldrich; EMD Millipore) and
SP600125 (8177; Cell Signaling Technology, Inc., Danvers, MA, USA)
were dissolved in dimethyl sulfoxide. In this study, 20 µM DHA, 100
µM PDTC, 10 µM JNK inhibitor SP600125 and 10 µM anisomycin, were
used to treat the cells.
Western blotting
Cells were washed with cold PBS and lysed in
radioimmunoprecipitation assay buffer (20 mM Tris pH 7.5, 150 mM
NaCl, 50 mM NaF, 1% NP40, 0.1% DOC, 0.1% SDS, 1 mM EDTA, 1 mM
phenylmethylsulfonyl fluoride and 1 µg/ml leupeptin). Protein
concentrations of the cell lysates were determined using the
bicinchoninic acid assay (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). Equal amounts of the protein (25 µg) from each sample were
separated by SDS-PAGE (7.5% polyacrylamide gel) and transferred on
to a PVDF membrane, which was blocked with 2.5% non-fat milk and
incubated overnight with primary antibody in PBS-T at 4°C.
Immunoreactivity was detected with horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G secondary
antibody (1:2,000; 7074; Cell Signaling Technology, Inc.) for 30
min at room temperature, and visualized with enhanced
chemiluminescence (Pierce ECL; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The primary antibodies included rabbit
anti-phospho-SAPK/JNK (Thr183/Tyr185) (1:1,000; 81E11), rabbit
anti-SAPK/JNK (1:2,000; 9252), mouse anti-inhibitor of kappa B
(IκB)-α (1:1,000; L35A5; Cell Signaling Technology, Inc.) and
anti-β-actin antibodies (1:5,000; A2228; Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany). The densitometry analyses were
performed using ImageJ software (NIH, Bethesda, MD).
Reverse transcription-quantitative
polymerase chain reaction
Total RNA from the cells was extracted using the
RNeasy Mini kit (Qiagen GmbH, Hilden, Germany) and cDNA was
synthesized using High Capacity RNA-to-cDNA Master mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). RT-qPCR was performed
using SYBR Green Master Mix (Thermo Fisher Scientific, Inc.) and a
ViiA7 Real-Time PCR system (Thermo Fisher Scientific, Inc.) with
the following cycling conditions: 50°C for 2 min and 95°C for 2
min, followed by 40 cycles of 95°C for 15 sec and 58°C for 1 min..
All PCR reactions were repeated in triplicate. Relative expression
was calculated using GAPDH as an internal control and calculated
following the ΔΔCq method (32). The
primer sequences are summarized in Table
I.
 | Table I.Reverse-transcription polymerase
chain reaction primer sequences. |
Table I.
Reverse-transcription polymerase
chain reaction primer sequences.
| Gene | Sequence | Size, bp | Tm, °C |
|---|
|
Cyclooxygenase-2 |
| 67 | 58.5 |
|
Sense |
GAATCATTCACCAGGCAAATTG |
|
|
|
Anti-sense |
TCTGTACTGCGGGTGGAACA |
|
|
| Matrix
metalloproteinase 13 |
| 123 | 59 |
|
Sense |
TCCCAGGAATTGGTGATAAAGTAGA |
|
|
|
Anti-sense |
CTGGCATGACGCGAACAATA |
|
|
| GAPDH |
| 240 | 63.5 |
|
Sense |
TGATGACATCAAGAAGGTGGTGAAG |
|
|
|
Anti-sense |
TCCTTGGAGGCCATGTGGGCCAT |
|
|
Caspase activity assay
The bioactivity of caspase-3 and −9 was measured
with a Fluorometric Assay kit (Abcam, Cambridge, UK) according to
the manufacturer's protocol. Briefly, the cells were collected
after 20 µM DHA treatment for 24 h and incubated with the caspase-3
substrate DEVD-AFC or the caspase-9 substrate LEHD-AFC. The
fluorescence of the cleaved substrates was determined at an
excitation wavelength of 400 nm and an emission wavelength of 505
nm using a fluorescence plate reader (Molecular Devices, LLC,
Sunnyvale, CA, USA).
Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) analyses
The apoptosis of HUVECs treated with DHA and/or the
inhibitors was detected by annexin V-FITC and propidium iodide (PI)
staining according to the manufacturer's protocol of the Apoptosis
Detection kit (Neobiosciences, Shenzhen, China). Briefly, the cells
were collected by trypsinization and resuspended in binding buffer
containing annexin V-FITC (0.25%) and PI (1 µg/ml). Detection of
the cells with positive staining was performed using a FACSAria II
flow cytometer (BD Biosciences, San Jose, CA, USA). The data were
analyzed with the FACS Diva acquisition and analysis software
version 11.5 (BD Biosciences).
Statistical analysis
Data were expressed as the mean ± standard error.
Comparison of means was achieved by the unpaired, two-tailed
Student's t-test. Statistical analyses were performed using SPSS
version 11.5 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
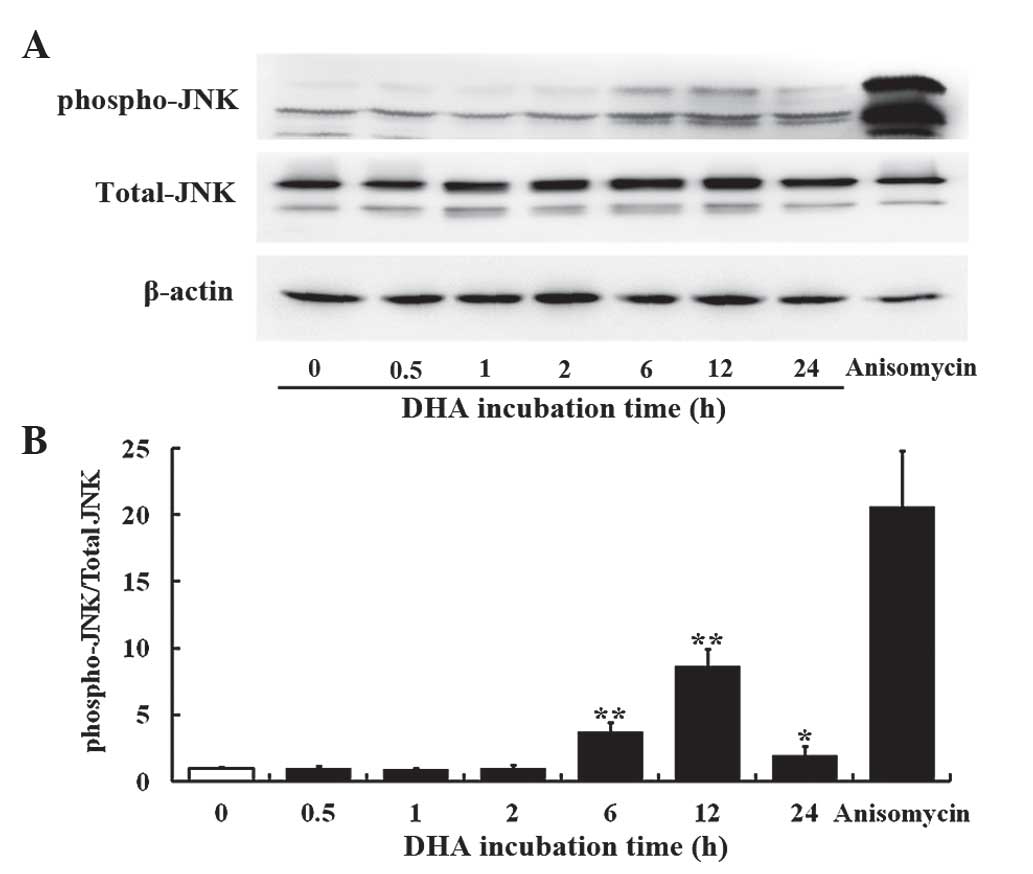
DHA transiently activates JNK in
endothelial cells
The JNK signaling pathway mediates cellular
responses to extracellular stimuli and is involved in angiogenesis
(14). The present study investigated
the effects of 20 µM DHA on phosphorylation of JNK in HUVECs by
western blotting. The protein levels of the total JNK remained
unaffected at all time points; however, the phospho-JNK level was
elevated at 6, 12 and 24 h incubation with DHA (Fig. 1A). Densitometry analysis revealed that
the ratio of phospho-JNK/total-JNK was significantly increased at 6
h (P=0.001), reached a peak at 12 h (P=0.003) and subsequently
decreased from the peak at 24 h (P=0.020) following DHA treatment
(Fig. 1B), suggesting that DHA
transiently activates the JNK signaling pathway. Anisomycin is a
phenylpyrolidine derivative that strongly activates JNK (33); thus, it was used as a positive control
in the present study. Anisomycin markedly increased the level of
phospho-JNK in HUVECs (Fig. 1A and
B).
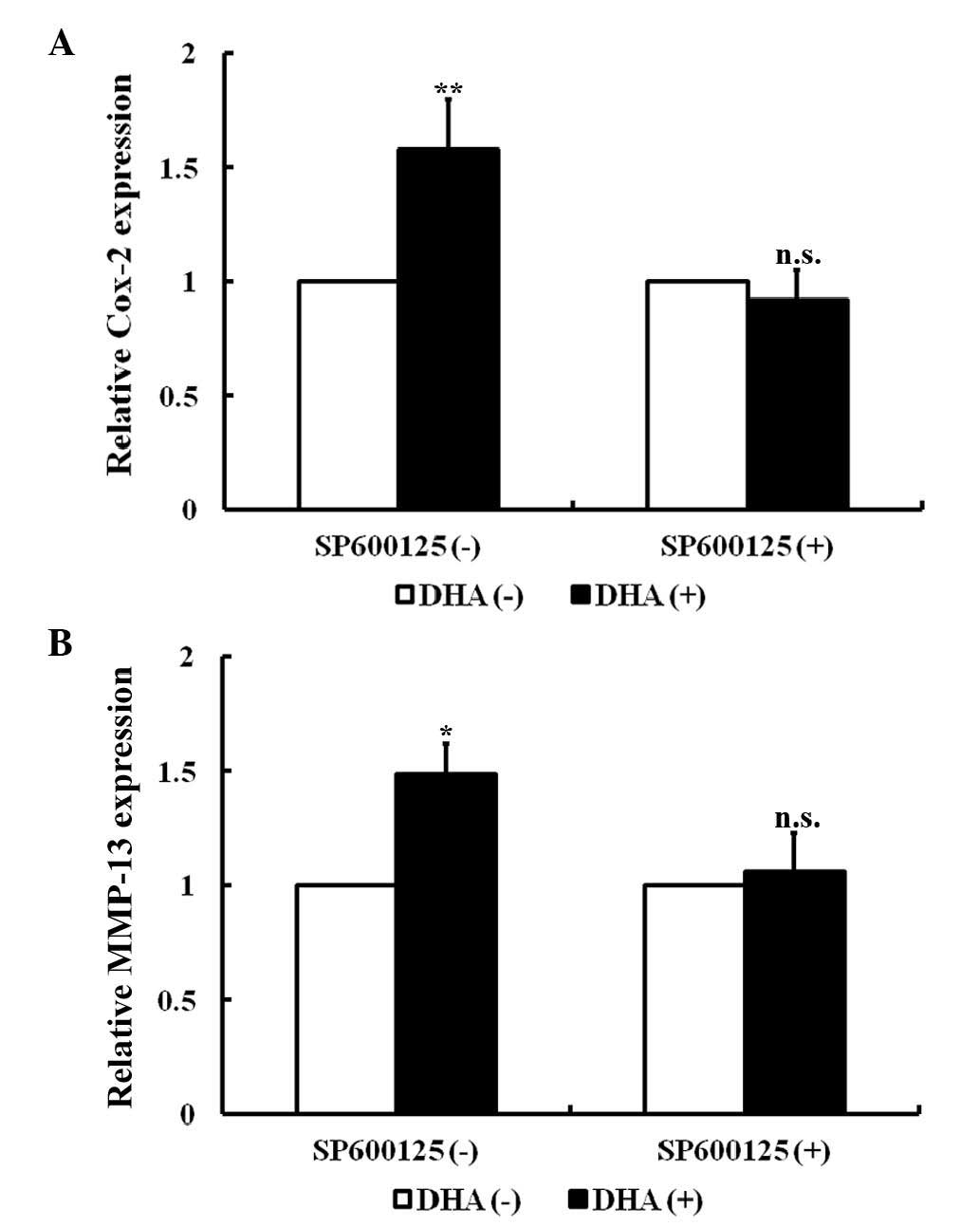
DHA induces the expression of the
downstream genes of the JNK signaling pathway in endothelial
cells
In endothelial cells, the JNK signaling pathway
specifically mediates the expression of cyclooxygenase-2 (Cox-2)
and matrix metalloproteinase-13 (MMP-13) in response to a variety
of cytokines (34,35). As revealed by RT-PCR, Cox-2 and MMP-13
transcription was significantly increased in HUVECs following
treatment with 20 µM DHA for 24 h (P=0.010 and P=0.020,
respectively; Fig. 2A and B).
SP600125 is an anthrapyrazolone inhibitor of JNK (36). With pretreatment of 10 µM SP600125 for
1 h, DHA failed to increase the mRNA levels of Cox-2 and MMP-13
(P=0.08 and P=0.11, respectively) (Fig.
2A and B). Thus, DHA induces expression of Cox-2 and MMP-13 via
transient activation of the JNK signaling pathway.
DHA does not induce JNK-mediated
apoptosis in endothelial cells
Apoptosis, a type of programmed cell death, is a
critical process that is altered during angiogenesis (37). JNK signaling activates intrinsic
apoptotic signaling pathways by modulating the activities of pro-
and anti-apoptotic proteins in mitochondria through phosphorylation
(16). The present study investigated
the effects of low dose DHA (20 µM) on endothelial cell apoptosis
by flow cytometry. As shown by Fig.
3A, DHA treatment does not alter the percentage of viable cells
(93.4±2.2 vs. 93.8±3.1; P=0.33) (Fig.
3A). Following pretreatment with 10 µM JNK inhibitor SP600125,
the percentage of viable cells remained unaffected by DHA treatment
(91.3±2.7 vs. 90.8±1.6; P=0.09) (Fig.
3B). JNK may increase caspase 3 and 9 activities to facilitate
apoptosis (38). However, treatment
with 20 µm DHA does not alter the activities of caspase 3 and 9 in
HUVECs (P=0.21 and P=0.18, respectively) (Fig. 3C). Taken together, the results of the
present study appear to indicate that low dose DHA does not affect
apoptosis in endothelial cells.
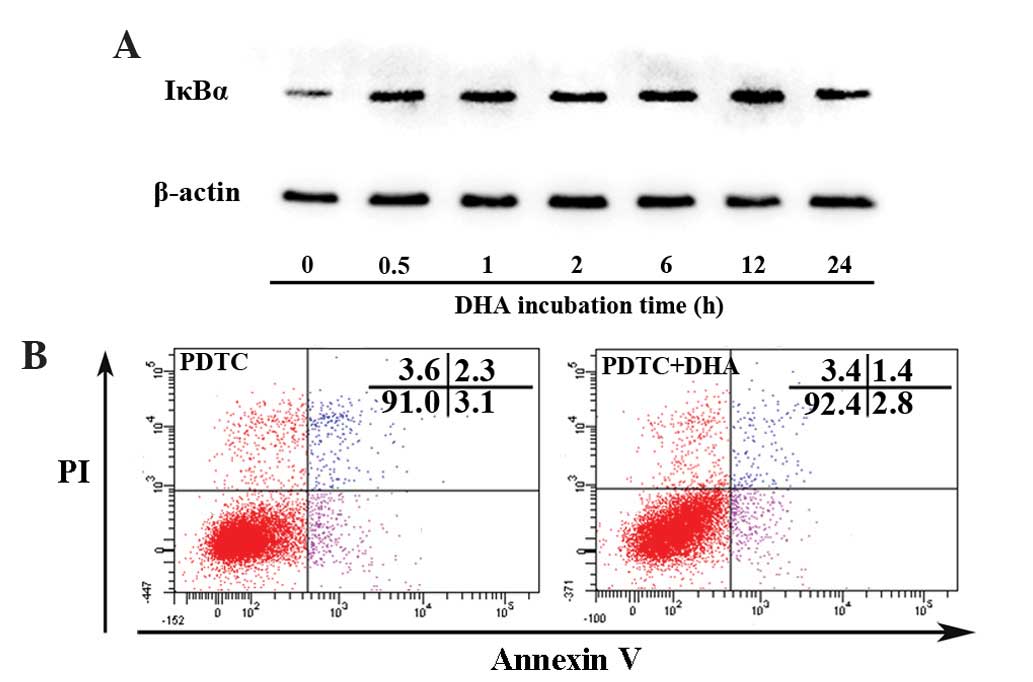
DHA persistently inhibits the NF-κB
signaling pathway in endothelial cells
Nuclear factor-κB (NF-κB) signaling regulates
expression of a large number of genes that are critical for the
regulation of apoptosis, which is mediated in part by its ability
to downregulate JNK activation (39).
Activation of NF-κB requires the degradation of IκB-α, which binds
with the p65-p50 heterodimer and blocks the nuclear translocation
of the NF-κB subunits (39). The
present study assessed whether DHA affects IκB-α by western
blotting. As shown in Fig. 4A, the
protein level of IκB-α began to increase at 0.5 h and peaked at 12
h following treatment with 20 µM DHA. PDTC is a potent inhibitor of
the NF-κB signaling pathway that inhibits IκB-α degradation,
precludes the dissociation of NF-κB from IκB-α and thus prevents
translocation of NF-κB to the nucleus (40). Following pretreatment with 100 µM PDTC
for 1 h, DHA showed no effect on apoptosis as determined by the
percentage of viable cells (91.1±1.9 vs. 91.6±2.5; P=0.17)
(Fig. 4B). Though DHA inhibits the
NF-κB signaling pathway by upregulation of IκB-α, it does not
affect apoptosis via NF-κB dependent or independent signaling
pathways.
Discussion
The artemisinin family of drugs demonstrates potent
inhibitory effects on angiogenesis; however, the underlying
molecular mechanisms remain unclear. In the present study, the role
of DHA, a water-soluble metabolite of artemisinin derivatives, on
the JNK signaling in endothelial cells was examined. It was
observed that DHA transiently activates JNK and upregulates the
expression of JNK signaling pathway downstream genes, which can be
abolished by JNK inhibitor SP600125. In addition, DHA does not
affect apoptosis or caspase activity despite persistently
inhibiting the NF-κB signaling pathway.
Angiogenesis is a complex process regulated by
multiple signaling pathways (37).
Many of the angiogenic pathways overlap, resulting in redundancy or
contradiction within the angiogenic system (37). MAPK cascades are involved in mediating
the effects of angiogenic and anti-angiogenic factors (41,42). It
has previously been reported that DHA inhibits endothelial cell
proliferation by suppression of ERK signaling (30), while it does not alter p38 MAPK
(31). In the present study, it was
observed that DHA activates transient but not prolonged JNK
activation in HUVECs. The expression of Cox-2 and MMP13, the
downstream genes of JNK activation in endothelial cells, were also
upregulated by DHA. Thus, DHA acted distinctly on the three major
MAPK signaling pathways in endothelial cells. The differential
regulation of MAPK cascades was also observed in other stress
responses (43). JNK signaling is a
positive and negative regulator of angiogenesis (44). Inhibition of JNK reduced endothelial
cell proliferation, migration and proteolysis of the capillary
basement membrane (45). By contrast,
the JNK signaling pathway mediates the anti-angiogenesis effects of
various agents by increasing endothelial cell apoptosis (46). Delineation of the effects of DHA on
specific signaling pathways may assist with the use of this agent
through combination therapy.
Activation of JNK promotes apoptosis in a
context-specific fashion (13).
Studies using deficient or constitutively active components of the
JNK signaling pathway indicate that activation of JNK increases
apoptosis (47,48). However, the pro-apoptotic role of the
JNK signaling pathway depends on the experimental settings. Under
certain circumstances, JNK signaling also serves an anti-apoptotic
role (49) or has no effects on
apoptosis (50). Though DHA activates
transient JNK activation, it was observed that DHA does not inhibit
endothelial cell apoptosis. In addition, DHA has no effect on the
activities of caspase 3 and 9, which is activated by the JNK
signaling pathway and mediates the JNK-dependent mitochondrial
apoptotic signaling pathway (38).
The effects of JNK activation on apoptosis were distinguished by
the varying activation patterns, transient vs.
prolonged/persistent, respectively. Previous studies demonstrated
that prolonged activation of JNK promotes tumor necrosis factor
(TNF)-α-induced apoptosis, but conversion of JNK activation from
prolonged to transient suppresses TNF-α-induced apoptosis (51). The effects of transient JNK activation
alone on apoptosis remain controversial, while various stimuli
induced transient JNK activation without causing apoptotic cell
death (52).
The NF-κB signaling pathway is a negative regulator
of apoptosis, and its anti-apoptotic function is mediated in part
through downregulation of JNK activation (52). DHA induced a sustained increase of
IκB, indicating its inhibitory role on NF-κB signaling. Thus, the
transient action of JNK may be caused by the inhibitory effects of
DHA on NF-κB signaling. However, DHA does not affect apoptosis in
the absence or presence of NF-κB inhibitor. In fibroblasts,
suppression of NF-κB induces prolonged (rather than transient) JNK
activation, leading to increased apoptosis (53). However, the crosstalk between NF-κB
and JNK is complicated (52). In
TNF-α-treated NF-κB-deficient cells, persistent JNK activation
promotes cell survival (54). In the
present study, DHA showed no significant effect on apoptosis
although it inhibited NF-κB and transiently activated JNK
signaling. Thus, NF-κB-regulated activation of JNK regulates
apoptosis based on death stimulus and cell types.
In summary, the present study observed that low
concentration of DHA induces transient activation of JNK signaling
without triggering apoptosis in endothelial cells. The results of
the present study provide important information concerning the
molecular mechanisms that underlie the anti-angiogenic activities
of DHA, which is essential for investigating its potential for
clinical application.
Acknowledgements
The present study was supported by grants from the
Science and Technology Development Plan of Jinan City (grant no.
201303026) and the Medical Science Development Plan of Shandong
Province (grant no. 2013WS0137. The authors are grateful for the
support from the Shandong Taishan Scholarship (awarded to Professor
Ju Liu).
References
|
1
|
Carmeliet P: Angiogenesis in life, disease
and medicine. Nature. 438:932–936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cao Y: Antiangiogenic cancer therapy: Why
do mouse and human patients respond in a different way to the same
drug? Int J Dev Biol. 55:557–562. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferrara N and Kerbel RS: Angiogenesis as a
therapeutic target. Nature. 438:967–974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bergers G and Hanahan D: Modes of
resistance to anti-angiogenic therapy. Nat Rev Cancer. 8:592–603.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ellis LM and Hicklin DJ: Pathways
mediating resistance to vascular endothelial growth factor-targeted
therapy. Clin Cancer Res. 14:6371–6375. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen HX and Cleck JN: Adverse effects of
anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol.
6:465–477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Folkman J: Angiogenesis in cancer,
vascular, rheumatoid and other disease. Nat Med. 1:27–31. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoefen RJ and Berk BC: The role of MAP
kinases in endothelial activation. Vascul Pharmacol. 38:271–273.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Page C and Doubell AF: Mitogen-activated
protein kinase (MAPK) in cardiac tissues. Mol Cell Biochem.
157:49–57. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shaulian E and Karin M: AP-1 as a
regulator of cell life and death. Nat Cell Biol. 4:E131–E136. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin A: Activation of the JNK signaling
pathway: Breaking the brake on apoptosis. Bioessays. 25:17–24.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Genet Dev. 12:14–21. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hibi M, Lin A, Smeal T, Minden A and Karin
M: Identification of an oncoprotein- and UV-responsive protein
kinase that binds and potentiates the c-Jun activation domain.
Genes Dev. 7:2135–2148. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J and Lin A: Role of JNK activation in
apoptosis: A double-edged sword. Cell Res. 15:36–42. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang J, Guo L, Zhou X, Dong F, Li L,
Cheng Z, Xu Y, Liang J, Xie Q and Liu J: Dihydroartemisinin induces
endothelial cell anoikis through the activation of the JNK
signaling pathway. Oncol Lett. 12:1896–1900. 2016.PubMed/NCBI
|
|
18
|
Kang YJ, Jeon ES, Song HY, Woo JS, Jung
JS, Kim YK and Kim JH: Role of c-Jun N-terminal kinase in the
PDGF-induced proliferation and migration of human adipose
tissue-derived mesenchymal stem cells. J Cell Biochem.
95:1135–1145. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng YL, Choi Y, Seow WL, Manzanero S,
Sobey CG, Jo DG and Arumugam TV: Evidence that neuronal Notch-1
promotes JNK/c-Jun activation and cell death following ischemic
stress. Brain Res. 1586:193–202. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tu Y: The development of new antimalarial
drugs: Qinghaosu and dihydro-qinghaosu. Chin Med J (Engl).
112:976–977. 1999.PubMed/NCBI
|
|
21
|
White NJ: Qinghaosu (artemisinin): The
price of success. Science. 320:330–334. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Hensbroek MB, Onyiorah E, Jaffar S,
Schneider G, Palmer A, Frenkel J, Enwere G, Forck S, Nusmeijer A,
Bennett S, et al: A trial of artemether or quinine in children with
cerebral malaria. N Engl J Med. 335:69–75. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen HH, Zhou HJ, Wang WQ and Wu GD:
Antimalarial dihydroartemisinin also inhibits angiogenesis. Cancer
Chemother Pharmacol. 53:423–432. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dong F, Zhou X, Li C, Yan S, Deng X, Cao
Z, Li L, Tang B, Allen TD and Liu J: Dihydroartemisinin targets
VEGFR2 via the NF-kB pathway in endothelial cells to inhibit
angiogenesis. Cancer Biol Ther. 15:1479–1488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou HJ, Wang WQ, Wu GD, Lee J and Li A:
Artesunate inhibits angiogenesis and downregulates vascular
endothelial growth factor expression in chronic myeloid leukemia
K562 cells. Vascul Pharmacol. 47:131–138. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen HH, Zhou HJ and Fang X: Inhibition of
human cancer cell line growth and human umbilical vein endothelial
cell angiogenesis by artemisinin derivatives in vitro. Pharmacol
Res. 48:231–236. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu GD, Zhou HJ and Wu XH: Apoptosis of
human umbilical vein endothelial cells induced by artesunate.
Vascul Pharmacol. 41:205–212. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
D'Alessandro S, Basilico N, Corbett Y,
Scaccabarozzi D, Omodeo-Salè F, Saresella M, Marventano I, Vaillant
M, Olliaro P and Taramelli D: Hypoxia modulates the effect of
dihydroartemisinin on endothelial cells. Biochem Pharmacol.
82:476–484. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ho WE, Peh HY, Chan TK and Wong WS:
Artemisinins: Pharmacological actions beyond anti-malarial.
Pharmacol Ther. 142:126–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dong F, Tian H, Yan S, Li L, Dong X, Wang
F, Li J, Li C, Cao Z, Liu X and Liu J: Dihydroartemisinin inhibits
endothelial cell proliferation through the suppression of the ERK
signaling pathway. Int J Mol Med. 35:1381–1387. 2015.PubMed/NCBI
|
|
31
|
Guo L, Dong F, Hou Y, Cai W, Zhou X, Huang
AL, Yang M, Allen TD and Liu J: Dihydroartemisinin inhibits
vascular endothelial growth factor-induced endothelial cell
migration by a p38 mitogen-activated protein kinase-independent
pathway. Exp Ther Med. 8:1707–1712. 2014.PubMed/NCBI
|
|
32
|
Pinent M, Hackl H, Burkard TR, Prokesch A,
Papak C, Scheideler M, Hämmerle G, Zechner R, Trajanoski Z and
Strauss JG: Differential transcriptional modulation of biological
processes in adipocyte triglyceride lipase and hormone-sensitive
lipase-deficient mice. Genomics. 92:26–32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cano E, Hazzalin CA and Mahadevan LC:
Anisomycin-activated protein kinases p45 and p55 but not
mitogen-activated protein kinases ERK-1 and −2 are implicated in
the induction of c-fos and c-jun. Mol Cell Biol. 14:7352–7362.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Z, Meng D, Li G, Xu J, Tian K and Li Y:
Celecoxib combined with diacerein effectively alleviates
osteoarthritis in rats via regulating JNK and p38MAPK signaling
pathways. Inflammation. 38:1563–1572. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kohlstedt K, Busse R and Fleming I:
Signaling via the angiotensin-converting enzyme enhances the
expression of cyclooxygenase-2 in endothelial cells. Hypertension.
45:126–132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cho SG and Choi EJ: Apoptotic signaling
pathways: Caspases and stress-activated protein kinases. J Biochem
Mol Biol. 35:24–27. 2002.PubMed/NCBI
|
|
39
|
Bubici C, Papa S, Pham CG, Zazzeroni F and
Franzoso G: The NF-kappaB-mediated control of ROS and JNK
signaling. Histol Histopathol. 21:69–80. 2006.PubMed/NCBI
|
|
40
|
Liu SF, Ye X and Malik AB: Pyrrolidine
dithiocarbamate prevents I-kappaB degradation and reduces
microvascular injury induced by lipopolysaccharide in multiple
organs. Mol Pharmacol. 55:658–667. 1999.PubMed/NCBI
|
|
41
|
Harris VK, Coticchia CM, Kagan BL, Ahmad
S, Wellstein A and Riegel AT: Induction of the angiogenic modulator
fibroblast growth factor-binding protein by epidermal growth factor
is mediated through both MEK/ERK and p38 signal transduction
pathways. J Biol Chem. 275:10802–10811. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huynh-Do U, Vindis C, Liu H, Cerretti DP,
McGrew JT, Enriquez M, Chen J and Daniel TO: Ephrin-B1 transduces
signals to activate integrin-mediated migration, attachment and
angiogenesis. J Cell Sci. 115:3073–3081. 2002.PubMed/NCBI
|
|
43
|
Liu J and Kapron CM: Differential
induction of MAP kinase signalling pathways by cadmium in primary
cultures of mouse embryo limb bud cells. Reprod Toxicol.
29:286–291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sakurai T and Kudo M: Signaling pathways
governing tumor angiogenesis. Oncology. 81:(Suppl 1). S24–S29.
2011. View Article : Google Scholar
|
|
45
|
Uchida C, Gee E, Ispanovic E and Haas TL:
JNK as a positive regulator of angiogenic potential in endothelial
cells. Cell Biol Int. 32:769–776. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Aggarwal BB: Tumour necrosis factors
receptor associated signalling molecules and their role in
activation of apoptosis, JNK and NF-kappaB. Ann Rheum Dis.
59:(Suppl 1). i6–i16. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ham J, Eilers A, Whitfield J, Neame SJ and
Shah B: c-Jun and the transcriptional control of neuronal
apoptosis. Biochem Pharmacol. 60:1015–1021. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lenczowski JM, Dominguez L, Eder AM, King
LB, Zacharchuk CM and Ashwell JD: Lack of a role for Jun kinase and
AP-1 in Fas-induced apoptosis. Mol Cell Biol. 17:170–181. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu ZG, Hsu H, Goeddel DV and Karin M:
Dissection of TNF receptor 1 effector functions: JNK activation is
not linked to apoptosis while NF-kappaB activation prevents cell
death. Cell. 87:565–576. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tang F, Tang G, Xiang J, Dai Q, Rosner MR
and Lin A: The absence of NF-kappaB-mediated inhibition of c-Jun
N-terminal kinase activation contributes to tumor necrosis factor
alpha-induced apoptosis. Mol Cell Biol. 22:8571–8579. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Papa S, Zazzeroni F, Pham CG, Bubici C and
Franzoso G: Linking JNK signaling to NF-kappaB: A key to survival.
J Cell Sci. 117:5197–5208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
De Smaele E, Zazzeroni F, Papa S, Nguyen
DU, Jin R, Jones J, Cong R and Franzoso G: Induction of gadd45beta
by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature.
414:308–313. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Reuther-Madrid JY, Kashatus D, Chen S, Li
X, Westwick J, Davis RJ, Earp HS, Wang CY and Baldwin AS Jr: The
p65/RelA subunit of NF-kappaB suppresses the sustained,
antiapoptotic activity of Jun kinase induced by tumor necrosis
factor. Mol Cell Biol. 22:8175–8183. 2002. View Article : Google Scholar : PubMed/NCBI
|