Introduction
Glioblastoma (GBM) is a devastating form of brain
cancer with a poor median survival time, high level of resistance
to current therapy and common recurrence following treatment
(1). The median survival time is 15
months, and this has improved little over the past four decades
(2). The current standard therapy for
GBM includes maximum debunking surgery, radiation and treatment
with the monofunctional alkylating agent temozolomide (TMZ), also
referred to as Temodar® (3). TMZ is a chemotherapeutic DNA-methylating
agent useful in the treatment of GBM (4). DNA O6-methylguanine (O6MG) lesions
created by drug exposure incorrectly pair with thymine during the
first cycle of DNA replication, leading to activation of the DNA
mismatch repair system (5). The
removal of the thymine opposite O6MG and subsequent reinsertion
following repair resynthesis leads to cycles of futile mismatch
repair (5). This ultimately leads to
DNA single-strand breaks, activation of sensors of DNA damage, the
generation of DNA double-strand breaks, and delayed cell death by a
combination of senescence and mitotic catastrophe (6,7). Despite
its ability to prolong disease-free survival, the toxicity of TMZ
treatment at typical doses has been proven to affect patient
quality of life (8,9). Thus, novel treatment options for
sensitizing GBM cells to TMZ chemotherapy are necessary.
Aurora-A is a serine/threonine kinase critical for
centrosome duplication, spindle assembly and mitotic exit (10–12). Human
Aurora-A maps to chromosomal region 20q13.2, which is amplified in
various types of cancer (12,13). Aurora-A is also overexpressed in terms
of mRNA and protein levels in various types of cancer. Ectopic
overexpression of Aurora-A has been shown to transform rodent cell
lines (14,15), establishing it as a proto-oncogene.
Aurora-A has been documented to be involved in p53 regulation as it
phosphorylates p53 at Ser315, leading to its mouse double minute
2-mediated ubiquitination and subsequent proteolysis (16). Aurora-A also drives cell cycle
progression by promoting cyclin B1, Wnt, myc and other
pro-proliferative signaling pathways (17–19).
Repression of Aurora-A by RNA interference delays tetraploidy,
apoptosis and senescence in several types of cancer cell (20,21).
In the present study, it was hypothesized that
knockdown of Aurora-A in GBM cells would greatly enhance their
sensitivity to TMZ. Thus, in vitro short hairpin (sh)RNA
targeting Aurora-A was demonstrated to be an appropriate tool to
knockdown Aurora-A expression at the protein and mRNA level in U251
cells. Subsequently, Cell Counting Kit-8 (CCK8) assays, flow
cytometric analysis, colony formation assays, invasion assays and
tube formation assays were performed and demonstrated that
knockdown of Aurora-A sensitizes GBM cells to TMZ in vitro.
Furthermore, TMZ treatment combined with knockdown of Aurora-A by
plasmid based shAurora-A/liposome complex significantly inhibited
U251 subcutaneous tumor growth, compared with TMZ treatment alone.
The results of the present study demonstrated that knockdown of
Aurora-A in GBM cells enhanced TMZ sensitivity in vitro and
in vivo. Aurora-A knockdown may be a novel treatment option
in order to decrease TMZ toxicity.
Materials and methods
Vector construction
Plasmids were constructed through usage of the
pGensil-2 parental vector (Genesil Biotechnology Co., Wuhan,
China). Oligonucleotide sequences of Aurora-A-shRNA
(CACCGATGCCCTGTCTTACTGTCATTCAAGAGATGACAGTAAGACAGGGCATTTTTTG) were
designed by Genesil Biotechnology Co. according to a published
sequence of Aurora-A-shRNA, which was shown to efficiently silence
Aurora-A expression in vitro and in vivo (22). Oligonucleotide sequences of
scramble-shRNA, which had no homology with any of the mammalian
sequence
(CACCGCGTACGCGGAATACTTCGATTCAAGAGATCGAAGTATTCCGCGTACGTTTTTG), were
designed as a negative control. The resulting recombinant plasmids
were named shAurora-A or shControl (shCtrl), respectively. The two
constructs were verified by DNA sequencing. Plasmids were extracted
using EndoFree Plasmid Giga kits (Qiagen GmbH, Hilden, Germany)
from DH5α Escherichia coli transformants and stored at −20°C
until use. The concentration was determined by measuring the
A260/A280 ratio using UV
spectrophotometry.
Cell line and treatment
The U251 and U87-MG cell lines were obtained from
American Type Culture Collection (Manassas, VA, USA). U251 and
U87-MG cells were cultured in Dulbecco's Modified Eagle's Medium
(DMEM) containing 10% fetal bovine serum (FBS) (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). All cells were
maintained in a humidified atmosphere containing 5% CO2
at 37°C. Cell transfection was performed using FuGENE®
HP Transfection Reagent (Roche Diagnostics, Indianapolis, IN, USA)
according to the manufacturer's protocol. Briefly, cells were
seeded into 6-well plates at a density of 2×105
cells/well and cultured for 24 h to reach 70–80% confluency. A
total of 2 µg plasmid was diluted in 100 µl media without serum and
5 µl FuGENE® HP Transfection Reagent was added to the
tubes containing the diluted DNA. These were subsequently mixed and
the transfection complex incubated for 15 min at room temperature,
before being added to the 6-well plates. Medium alone was used as
blank control. Simultaneously, cells were treated with a dose of 10
µM TMZ. Cells and cell supernatant were harvested 48 h subsequent
to transfection for reverse transcription-polymerase chain reaction
(RT-PCR) analysis, western blotting, colony formation assays, cell
proliferation assays and human umbilical vein endothelial cells
(HUVEC) tube formation analysis. All treatments were performed in
triplicate.
RNA extraction and RT-PCR
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RNA samples (1 µg) were subjected to reverse transcription
using the Takara Primescript RT-PCR kit (Takara Bio, Inc., Otsu,
Japan). The primer sequences used were as follows: Aurora-A (515
bp) 5′-GAGGCAGTGGGCTTTGG-3′ (sense) and 5′-GGCAGGTAGTCCAGGGTG-3′
(antisense). RT-PCR was performed with reverse transcription at
50°C for 30 min, followed by initial denaturation at 94°C for 3 min
and 30 cycles of 30 sec at 94°C, 30 sec at 60°C and 45 sec at 72°C.
All PCR products were separated by electrophoresis on 1% agarose
gels and visualized using ethidium bromide. The amplified products
were quantified by Quantity One software (Version 4.1; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Experiments were performed
in triplicate.
Western blotting
Cells were lysed on ice for 30 min with
radioimmunoprecipitation assay lysis buffer [containing 50 mM
Tris-HCl, pH 7.4; 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1
mM ethylenediaminetetraacetic acid (EDTA), 1 mM phenylmethane
sulfonyl fluoride, 1 µg/ml aprotinin, 1 µg/ml leupeptin, 1 µg/ml
pepstatin, 1 mM Na3VO4 and 1 mM NaF]. The
protein concentration was determined by the bicinchoninic acid
assay (Beyotime Institute of Biotechnology, Haimen, China), and the
proteins (25 µg) were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and electronically
transferred onto a polyvinylidene difluoride membrane (EMD
Millipore, Billerica, MA, USA). Following blocking in tris-buffered
saline and Tween 20 (TBST) buffer containing 5% milk, the membranes
were incubated with primary antibodies against Aurora-A (catalog
no., 14475; dilution, 1:500; Cell Signaling Technology, Inc.,
Danvers, MA, USA), p53 (catalog no., 2524; dilution, 1:1,200; Cell
Signaling Technology, Inc.), caspase 3 (catalog no., 9661;
dilution, 1:600; Cell Signaling Technology, Inc.), matrix
metalloproteinase-2 (MMP-2) (catalog no., 4022; dilution, 1:800;
Cell Signaling Technology, Inc.), MMP-9 (catalog no., 2270;
dilution, dilution, 1:1,000; Cell Signaling Technology, Inc.) and
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (catalog no.,
sc-25778; dilution, 1:5,000; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) at 4°C overnight, followed by washing with TBST
buffer and incubation with the following horseradish
peroxidase-conjugated secondary antibodies: Goat anti-mouse
immunoglobulin G (catalog no., ab97040; dilution, 1:5,000) or goat
anti-rabbit (catalog no., ab191866; dilution, 1:5,000; Abcam,
Cambridge, MA, USA) at 37°C for 1 h. Bands were visualized using an
enhanced chemiluminescence kit (EMD Millipore). The ratio of
Aurora-A/GAPDH was calculated by using densitometry (on Image J;
Version 1.0, National Institutes of Health, Bethseda, MD, USA) and
values were normalized by dividing by the ratio at blank (23).
Flow cytometric analysis (FCM)
FCM was performed to identify apoptosis-positive
cells by propidium iodide (PI) (Beyotime Institute of
Biotechnology) staining. Briefly, cells were harvested with
trypsin-EDTA, washed with phosphate buffered saline (PBS) twice,
and pelleted (by centrifugation at 1,200 × g for 3 min at
4°C) and suspended in 500 µl binding buffer. A total of 5 µl PI was
subsequently added. Following incubation at room temperature for 15
min, analysis was performed on a FACSCalibur system (BD
Biosciences, San Jose, CA, USA). Sub-G1 cells were classified as
apoptosis positive cells.
Cell proliferation assay
U251 and U87-MG cells were seeded into 96 well
plates at a density of 5×103 cells per well in 200 µl
medium. Cell proliferation was monitored using the CCK8 assay
(Dojindo Molecular Technologies, Inc., Kumamoto, Japan). Briefly,
10 µl CCK8 solution was added into each well, and 4 h later, the
absorbance was determined at a wavelength of 450 nm. Experiments
were performed in triplicate.
Colony formation assay
Transfected cells were seeded into 6-well plates at
a density of 1×103 cells per well. After 7 days of
incubation at 37°C the cells were fixed with 100% methanol, stained
with 0.05% crystal violet (Beyotime Institute of Biotechnology) and
washed with PBS. The colonies were counted with an Olympus BX600
microscope in at least six randomly chosen fields. The results of
three independent experiments are given.
In vitro tube formation assay
The wells of a 96-well plate were coated with
ice-cold Matrigel™ (BD Biosciences). Following polymerization of
the matrix at 37°C, HUVECs isolated in the method described by Dai
et al (24) were seeded onto
the polymerized extracellular matrix at a density of
2×104 cells in 100 µl of extracellular growth medium-2
(Lonza, Walkersville, MD, USA) per well; 100 µl of the cell
supernatant collected from the transfected cells was immediately
added. The tube branches were photographed using a Nikon Eclipse
Ti-U following 6 h of incubation. The capillary-like structures of
the HUVECs were counted in three randomly selected fields. The
results of three independent experiments are presented.
Invasion assay
The invasion of U251 cells was evaluated using
24-well Transwells® (8-lm poresize; EMD Millipore), as
described previously but with slight modification (25). Briefly, the filter of the Transwell
plate was coated with 50 µl Matrigel. Following Matrigel
polymerization, 500 µl DMEM containing 10% FBS was placed in the
lower chamber and 100 µl U251 suspension (2×104
cells/well; in DMEM without 10% FBS) was placed in the upper
chamber. Following incubation for 48 h, cells on the lower surface
were fixed in 100% methanol, stained with 0.05% crystal violet,
counted in at least six randomly selected fields and photographed
using an Olympus BX600 microscope.
Enzyme-linked immunosorbent assay
(ELISA)
A total of 48 h after transfection, cell
supernatants were collected and stored at −80°C. The supernatant
was used to measure the total levels of several cytokines using the
human vascular endothelial growth factor (VEGF) ELISA kit
(NeoBioscience, Shenzhen, China) according to the manufacturer's
protocol.
Cationic liposome and preparation of
plasmid DNA/lipoplexes
Cationic liposome was prepared as multilamellar
vesicles for in vivo use as described previously (24). Briefly, 1,2
dioleoyl-3-trimethylammonium-propoane (Avanti Polar Lipids, Inc.
Alabaster, AL, USA) and cholesterol (Sigma-Aldrich; EMD Millipore)
were mixed in a 1:1 molar ratio, dehydrated in round-bottom tubes
using a freeze dryer (SCIENTZ-50ND; Scientz Biotechnology Co.,
Ltd., Ningbo, China), then rehydrated in 5% glucose solution by
heating at 50°C for 6 h. For the in vivo injection, plasmid
DNA/lipoplexes were prepared immediately prior to injection by
gently mixing cationic liposome with plasmid DNA at a ratio of 12.5
µg total cationic liposome to 2.5 µg plasmid DNA, resulting in a
final concentration of 12.5 µg plasmid DNA per ml in a sterile
solution of 5% glucose in water.
Tumor growth and treatments in
vivo
To establish a U251 subcutaneous cancer model, nude
female BALB/c mice (6–8 weeks old; 18–20 g weight) were obtained
from The Animal Center of Sichuan University (Chengdu, China) and
housed in a 26°C and 50% humidity-controlled facility, with a 24 h
light/dark cycle and free access to food and water. A total of
5×106 U251 cells were injected subcutaneously into the
right flank regions of nude female BALB/c mice (6–8 weeks old).
After one week, when the tumors were palpable, the animals were
randomly assigned to five independent groups, each group containing
five mice, and subjected to systemic treatment with one of the
following: shCtrl, dimethyl sulfoxide (DMSO), shAurora-A, TMZ
(Sigma-Aldrich; EMD Millipore) and shAurora-A+TMZ. Plasmid DNA (2.5
µg) and cationic liposome (12.5 µg), in 100 µl of 5% glucose was
injected into the mouse tail vein three times per week (on Mondays,
Wednesdays and Fridays). TMZ was intraperitoneally injected once
every 3 days at a dose of 200 mg/kg. Tumor diameters were measured
once every three days during the treatment period. Tumor volume was
estimated using the following formula: Tumor volume
(mm3) = length(mm) × [width(mm)]2 x 1/2. The
weight, appetite and behavior of the mice were observed. Mice were
sacrificed through cervical dislocation following 10 series of
treatment and dissected tumors were weighed. Animal studies were
performed in accordance with the Institutional Animal Care and
Treatment Committee of Sichuan University (Chengdu, China), and
written ethical approval was also provided by this board.
Immunostaining
Expression of cluster of differentiation 31 (CD31)
was measured with a rat anti-mouse CD31 antibody (catalog no.,
550674; BD Biosciences). Tumor sections (3–5 µm) of frozen tissues
were mounted on 3-aminopropyl triethoxysilane-coated glass slides.
Sections were fixed with 4% paraformaldehyde and washed with PBS
(pH 7.4). Endogenous peroxide was blocked with 3%
H2O2 for 10 min. Following PBS washes, slides
were blocked with 5% normal goat serum (Beijing Zhongshan Golden
Bridge Biotechnology Co., Ltd., Beijing, China) in PBS for 15 min
at room temperature, followed by incubation with primary anti-CD31
(1:400) antibody in blocking solution overnight at 4°C. All slides
were subsequently incubated with a 1:200 dilution of
biotin-conjugated goat anti-rat secondary antibody (catalog no.,
PV6004; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.)
for 15 min at 37°C and streptavidin-biotin complex at 37°C for 15
min. The immunoreaction was visualized using diaminobenzidine
peroxide solution (Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd.) and cellular nuclei were counterstained with hematoxylin
(Beyotime Institute of Biotechnology). All specimens were evaluated
using the Olympus BX600 microscope and Spot Fiex camera. Control
samples exposed to secondary antibody alone exhibited no specific
staining.
TUNEL
To detect apoptotic cells in tumor tissue, the TUNEL
assay was performed according to the manufacturer's protocol using
a DeadEnd™ Fluorometric TUNEL System (Promega Corporation, Madison,
WI, USA). Cell nuclei with dark green fluorescent staining were
defined as TUNEL-positive. TUNEL-positive nuclei were monitored by
fluorescence microscopy. To quantify TUNEL-positive cells, the
number of green fluorescence-positive cells was counted in random
fields at magnification, ×200.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical analysis was performed by Student's t-test for
comparing two groups and by analysis of variance for multiple group
comparisons with one-way analysis of variance. P<0.05 was
considered to indicate a statistically significant difference. SPSS
version 19 (IBM SPSS, Armonk, NY, USA) was used for all statistical
analyses.
Results
Knockdown of Aurora-A sensitizes
glioblastoma cells to TMZ in vitro
The shAurora-A and shCtrl plasmids were transfected
into U251 GBM cancer cells. A total of 48 h later, the cells were
harvested and the expression level of Aurora-A was analyzed by
western blotting and RT-PCR. As shown in Fig. 1A and B, suppression of Aurora-A
expression was observed in U251 cells transfected with shAurora-A
plasmid at the protein and mRNA level when compared with the
control group. Subsequently, the effect of shAurora-A combined with
TMZ on U251 and U87-MG cell proliferation was examined by CCK8
assay. The absorbance was determined at 0, 24, 48, 72 and 96 h
following U251 and U87-MG cell transfection and treatment with TMZ.
As shown in Fig. 1C and D, knockdown
of Aurora-A significantly inhibits U251 (P=0.0062) and U87-MG
(P=0.0072) cancer cell growth. Furthermore, TMZ treatment alone may
inhibit U251 and U87-MG cancer cell growth. In addition, the U251
(P=0.0042) and U87-MG (P=0.0059) cells that were transfected with
shAurora-A and treated with TMZ demonstrated significantly
increased inhibition of proliferation compared with the group
treated with TMZ alone. The above results indicate that knockdown
of Aurora-A sensitizes GBM cells to TMZ in vitro.
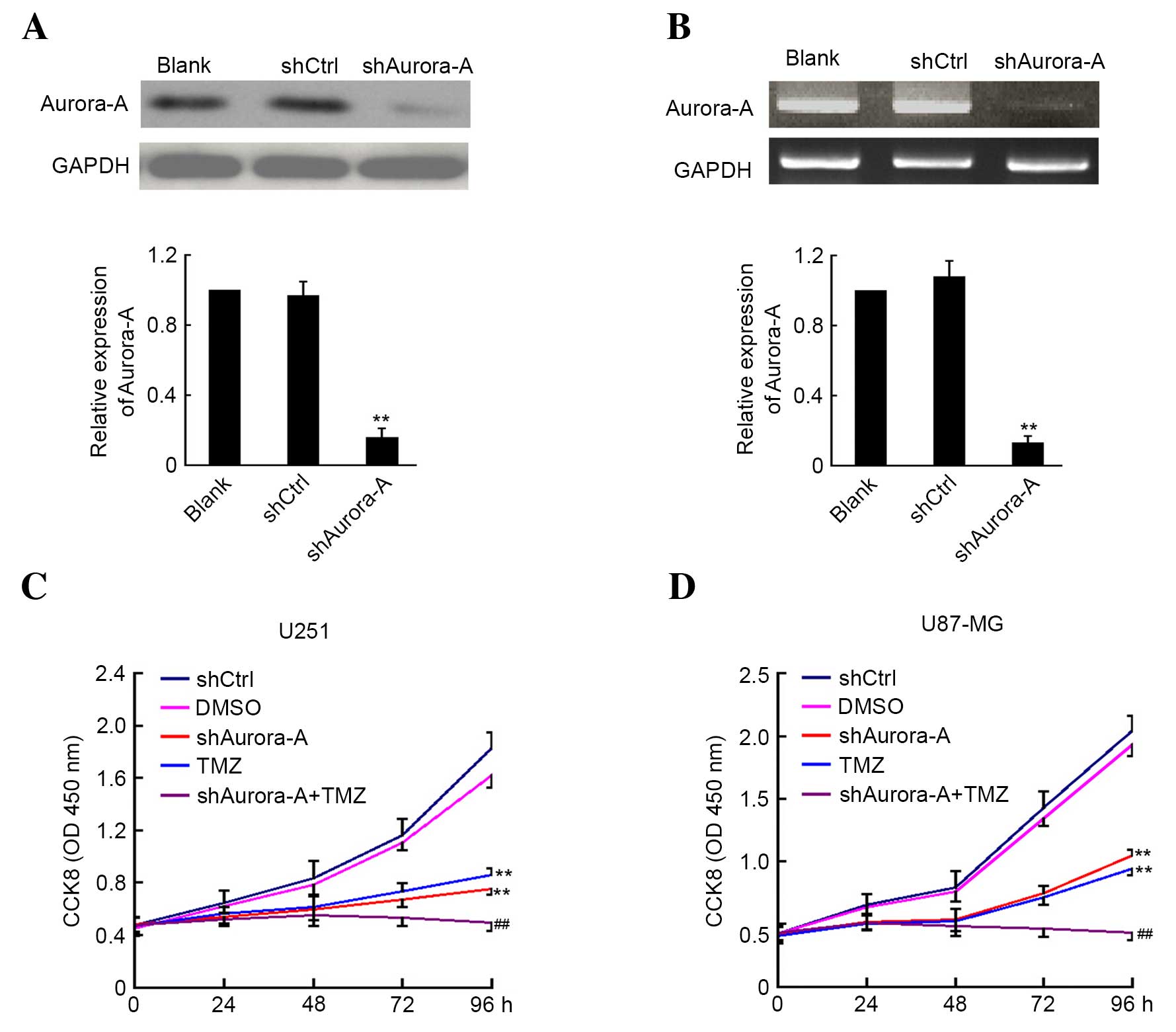 | Figure 1.Knockdown of Aurora-A sensitizes
glioblastoma cells to TMZ in vitro. U251 cells were
transfected with plasmid-based shAurora-A, shCtrl or medium alone
as control for 48 h. Aurora-A expression was detected by (A)
western blotting or (B) reverse transcription-polymerase chain
reaction analysis. GAPDH expression was monitored as the control.
The ratio of Aurora-A/GAPDH was calculated using densitometry. The
experiments were performed in triplicate and the mean ratio is
shown. The transfected (C) U251 and (D) U87-MG cells were plated
for CCK8 assay and treated with or without TMZ. DMSO was used as
the vehicle control. In this analysis, the absorbance was examined
at 450 nm 0, 24, 48, 72 and 96 h after cell plating (n=5). Values
are presented as the mean ± standard deviation. The experiment was
performed in triplicate. **P<0.01 compared with shCtrl group;
##P<0.01 compared with TMZ group. sh, short hairpin;
Ctrl, control; GAPDH; glyceraldehyde 3-phosphate dehydrogenase;
CCK8, cell counting Kit-8; TMZ, temozolomide; DMSO, dimethyl
sulfoxide; OD, optical density. |
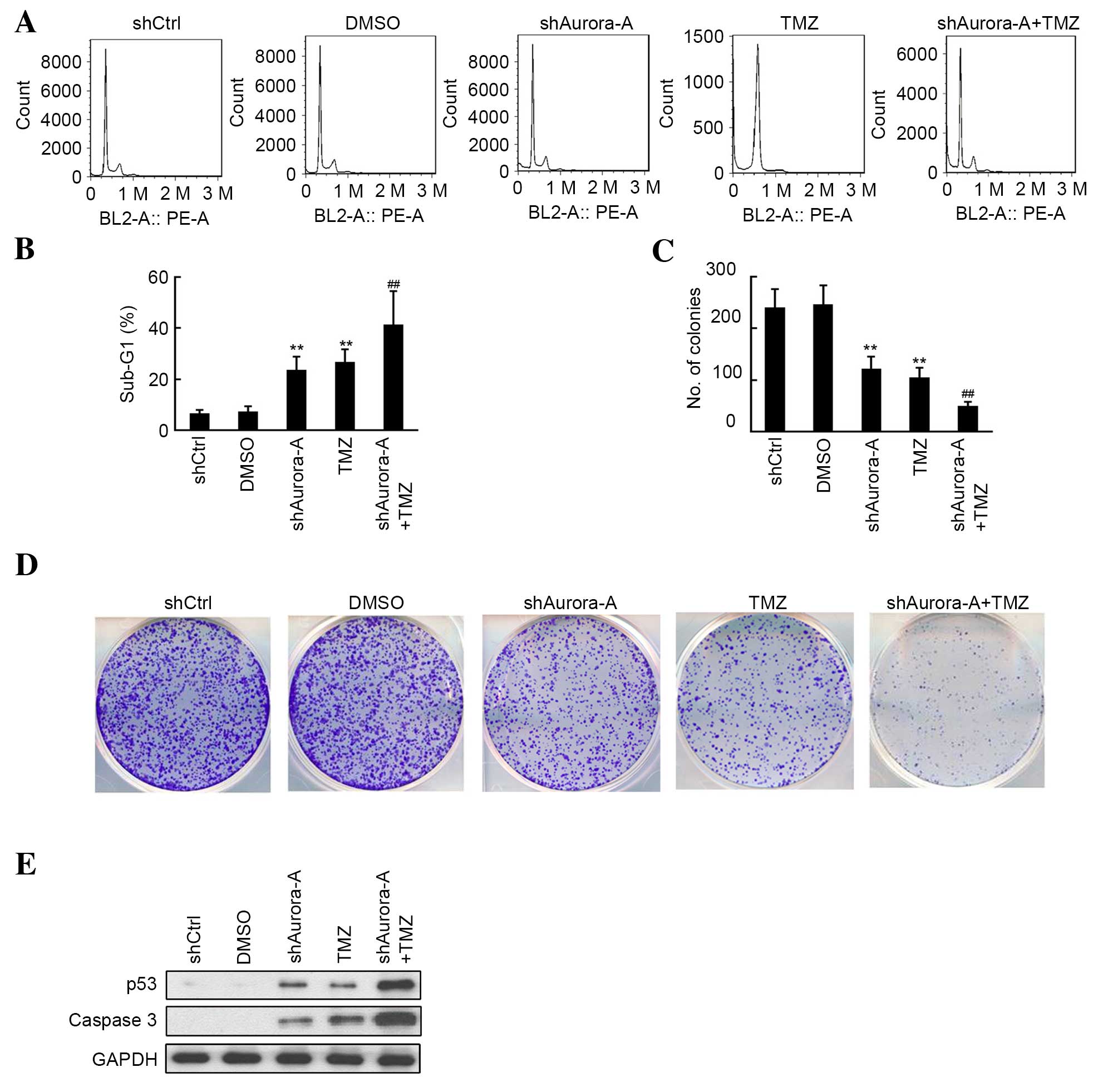
Knockdown of Aurora-A increases the
effects of TMZ treatment on GBM cell apoptosis and colony
formation
The quantitative assessment of sub-G1 cells by FCM
was used to estimate the number of apoptotic cells. As shown in
Fig. 2A and B, an increase in the
sub-G1 phase population was observed by flow cytometric analysis
following knockdown of Aurora-A or treatment with TMZ in U251 cell
lines. Furthermore, U251 cells transfected with shAurora-A
exhibited significantly increased apoptosis (P=0.0007) compared
with the TMZ-treated group. As shown in Fig. 2C and D, the inhibition of U251 cell
colony formation in the shAurora-A and TMZ-treated group was
measured, and compared with the shCtrl and DMSO groups,
respectively. Additionally, knockdown of Aurora-A increased the
inhibitive effect of TMZ treatment on U251 colony formation,
compared with the group treated with TMZ alone. The results of
western blot analysis shown in Fig.
2E indicated that shAurora-A (P=0.0038) or TMZ (P=0.0009)
treatment alone significantly induced the apoptosis-associated
protein p53, as well as caspase 3 expression. Furthermore,
increased p53 and caspase 3 expression was observed in the
shAurora-A+TMZ group. The above results indicate that knockdown of
Aurora-A increases the effects of TMZ on GBM cell apoptosis and
colony formation through induced p53 and caspase 3 expression.
Knockdown of Aurora-A increases the
effects of TMZ treatment on GBM cell invasion and angiogenesis
Invasion and tube formation assays were performed to
determine whether there is a synergistic effect in the group
knockdown of Aurora-A in U251 cells combined with TMZ treatment. As
shown in Fig. 3A and B, knockdown of
Aurora-A (P=0.0029) and TMZ treatment (P=0.023) alone significantly
inhibited U251 invasion, compared with the shCtrl or DMSO groups,
respectively. Furthermore, fewer invasive cells were observed in
the shAurora-A combined with TMZ treatment group, when compared
with the TMZ only group. Subsequently, western blotting results
indicated that MMP-2 and MMP-9 levels were inhibited following
shAurora-A transfection (Fig. 3C).
The conditional medium from each group was used to perform the
HUVEC tube formation assay. The results of the present study
demonstrated that the conditional medium from the shAurora-A
(P=0.0026) or TMZ (P=0.017) treated groups significantly inhibited
HUVEC tube formation, compared with the shCtrl or DMSO groups,
respectively (Fig. 3D and E).
Furthermore, knockdown of Aurora-A increased the inhibition of tube
formation in HUVECs, compared with the TMZ-treated group (Fig. 3D and E). Subsequently, VEGF expression
for each group was determined by ELIZA assay and decreased VEGF was
observed in the shAurora-A group compared with the shCtrl group. A
further decrease in VEGF expression was observed in the
shAurora-A+TMZ-treated group when compared with the TMZ-treated
alone group (Fig. 3D and E). The
above results demonstrate that knockdown of Aurora-A increases the
effects of TMZ treatment on GBM cell invasion and angiogenesis
through the inhibition of MMP-2, MMP-9 and VEGF expression.
 | Figure 3.Effects of shAurora-A on U251
invasion and tube formation. (A) Cell invasion in U251 cells was
evaluated using Transwell chambers. These experiments were
performed in triplicate (magnification, ×200). (B) The average
number of invasive cells is shown. Values are presented as the mean
± SD. **P<0.01 compared with shCtrl group; *P<0.05 compared
with shCtrl group; ##P<0.01 compared with TMZ group.
(C) Western blot analysis was performed to measure the expression
of the invasion-associated proteins MMP-2 and MMP-9 in each group.
(D) Conditional medium from each group was used for tube formation
assays in human umbilical vein endothelial cells. These experiments
were performed in triplicate (magnification, ×200). (E) The tubular
structure in each group was quantified by manual counting. The mean
number of branch points is shown. Values are presented as the mean
± SD. **P<0.01 compared with shCtrl group; *P<0.05 compared
with shCtrl group; ##P<0.01 compared with TMZ group.
(F) VEGF expression from the conditional medium of each group was
determined via enzyme-linked immunosorbent assay. These experiments
were performed in triplicate. The average concentrations of VEGF
are shown. Values are presented as the mean ± SD. **P<0.01
compared with shCtrl group; ##P<0.01 compared with
TMZ group. sh, short hairpin; SD, standard deviation; TMZ,
temozolomide; MMP, matrix metallopeptidase; Ctrl, control; DMSO,
dimethyl sulfoxide; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; VEGF, vascular endothelial growth factor. |
Knockdown of Aurora-A sensitizes GBM
cells to TMZ treatment in mice
The present study determined the effects of
silencing Aurora-A on tumor growth and in mice through
plasmid-based shRNA. A U251 GBM subcutaneous mouse model was used.
A plasmid/liposome complex was systemically injected through the
mouse tail vein, combined with or without TMZ treatment. As shown
in Fig. 4A, injection of the
shAurora-A/liposome complex significantly inhibited U251
subcutaneous growth, compared with the shCtrl/liposome complex
group (n=5, tumor volume: shAurora-A/liposome group, 1,063.4±79.3
mm3 vs. shCtrl/liposome group, 2,103.2±216.4
mm3; P=0.0051). Furthermore, TMZ treatment also caused
inhibition of U251 tumor growth compared with the DMSO group (n=5,
tumor volume: TMZ group, 894.6±68.9 mm3 vs. DMSO group,
2,275.4±221.4 mm3; P=0.0036). Notably the
shAurora-A/liposome complex combined with TMZ treatment
significantly inhibited U251 tumor growth compared with the TMZ
only group (n=5, tumor volume: Combined group, 422.6±48.3
mm3 vs. TMZ group, 894.6±68.9 mm3; P=0.0084).
Furthermore, U251 tumor weight was also inhibited by combined
shAurora-A/liposome complex and TMZ treatment, compared with
shCtrl/liposome and DMSO groups, respectively (Fig. 4B). The combined treatment group also
displayed markedly decreased tumor weight, when compared with the
TMZ only group (n=5, tumor weight: Combined group, 0.41±0.13 g vs.
TMZ group 0.91±0.18 g; P=0.0067; Fig.
4B).
 | Figure 4.Knockdown of Aurora-A sensitizes
glioblastoma cells to TMZ in mice. (A) Female nude mice at 6–8
weeks of age were implanted subcutaneously with U251 cells. A total
of 7 days after tumor cells were implanted, the mice were assigned
randomly to five groups and treated with an shAurora-A/liposome or
shCtrl/liposome complex, combined with or without TMZ. Tumor volume
was recorded during the treatment and growth curves were
constructed. Values are presented as the mean ± SD (n=5).
**P<0.01 compared with shCtrl group; ##P<0.01
compared with TMZ group. (B) A total of 3 days after the final
treatment, mice were sacrificed and subcutaneous tumors were
weighed. Values are presented as the mean ± SD (n=5). **P<0.01
compared with shCtrl group; ##P<0.01 compared with
TMZ group. (C and D) The TUNEL assay was performed to detect
apoptotic cells in primary U251 xenografts (magnification, ×100).
The apoptotic index was calculated as a ratio of the apoptotic cell
number to the total cell number in each field. Values are presented
as the mean ± SD (n=5). **P<0.01 compared with shCtrl group;
##P<0.01 compared with TMZ group. (E) Cluster of
differentiation 31 staining was performed to detect angiogenesis in
primary U251 xenografts (magnification, ×200). (F) The microvessel
density was calculated. Values are presented as the mean ± SD
(n=5). **P<0.01 compared with shCtrl group; *P<0.05 compared
with shCtrl group; ##P<0.01 compared with TMZ group.
TMZ, temozolomide; sh, short hairpin; SD, standard deviation; DMSO,
dimethyl sulfoxide; Ctrl, control. |
To detect apoptotic cells in tumor tissue, the TUNEL
assay was performed. As shown in Fig. 4C
and D, an increased number of apoptotic cells was observed in
the shAurora-A/liposome complex+TMZ group, compared with the
shCtrl/liposome complex or DMSO groups, respectively (n=5;
P=0.0052). Furthermore, there was an increased number of apoptotic
cells in the shAurora-A/liposome complex+TMZ group, compared with
the TMZ only group (n=5; P=0.0037). CD31 staining indicated that
fewer microvessels were observed in the shAurora-A/liposome
complex+TMZ group, compared with the shCtrl/liposome complex or
DMSO groups, respectively (n=5; P=0.0028; Fig. 4E and F). Furthermore, there were fewer
microvessels in the shAurora-A/liposome complex+TMZ group, compared
with the TMZ only group (n=5; P=0.0042; Fig. 4E and F). The above results indicate
that knockdown of Aurora-A sensitizes GBM cells to TMZ in mice by
inducing apoptosis and inhibiting angiogenesis.
Discussion
The present study demonstrates that the therapeutic
knockdown of Aurora-A through plasmid-based shAurora-A sensitizes
GBM cells to TMZ chemotherapy in vitro and in vivo.
The plasmid encoding shRNA targeting Aurora-A was proven to
knockdown Aurora-A expression at the mRNA and protein level in U251
cells. Furthermore, CCK8 assays, flow cytometric analysis, colony
formation assays, invasion assays and tube formation assays were
used to demonstrate that knockdown of Aurora-A sensitizes GBM cells
to TMZ in vitro. Notably, TMZ combined with knockdown of
Aurora-A through plasmid based shAurora-A/liposome complex
injection, significantly inhibited U251 subcutaneous tumor growth
compared with TMZ treatment alone.
Aurora-A is widely expressed in several types of
cancer and has been observed to be inversely correlated with
patient prognosis (14,15). Previous studies indicated that
Aurora-A is essential for the tumorigenic capacity and
chemoresistance of colorectal cancer stem cells (26). Thus, Aurora-A may be an effective
target for gene therapy and chemotherapy. A study by Van Brocklyn
et al (14) indicated that
knockdown of Aurora-A in GBM by alisertib (MLN8237), a highly
selective Aurora-A kinase inhibitor, extended the median survival
time of mice bearing intracranial human GBM neurosphere tumor
xenografts. However, whether knockdown of Aurora-A sensitizes GBM
cells to TMZ treatment remains to be elucidated. In the present
study, plasmid-based shRNA targeting Aurora-A was used to inhibit
its expression in GBM cells. The results of the present study
indicate that shAurora-A significantly inhibits the expression of
Aurora-A at the mRNA and protein level. Knockdown of Aurora-A may
significantly inhibit glioblastoma cell proliferation, colony
formation, invasion, angiogenesis and apoptosis induction in
vitro and in vivo. Further results demonstrate that
inhibition of Aurora-A in U251 cells induces expression of the
pro-apoptotic proteins p53 and caspase 3. Furthermore, MMP-2, MMP-9
and VEGF expression were decreased by shAurora-A.
TMZ is a widely used chemotherapeutic agent in the
treatment of GBM (4). Despite the
clinical occurrence of TMZ resistance (27), the dose of TMZ used significantly
affects the patient quality of life (8,9). Thus, in
recent years studies have focused on developing novel treatment
options to sensitize GBM cells to TMZ. Several inhibitors,
including KML001 and nelfinavir were proven to sensitize GBM cells
to TMZ treatment (28,29). In the present study, TMZ treatment
alone was proven to significantly inhibit glioblastoma cell
proliferation, colony formation, invasion, angiogenesis, and
apoptosis induction in vitro and in vivo. These
observations are consistent with the results of previous studies
(30,31). Notably, knockdown of Aurora-A in GBM
cells greatly enhances TMZ sensitivity in vitro and in
vivo, compared with TMZ treatment alone. This is due to the
altered expression of Aurora-A regulated proteins, including p53,
caspase 3, MMP-2, MMP-9 and VEGF.
The results of the present study demonstrate that
knockdown of Aurora-A in GBM cells greatly enhances TMZ sensitivity
in vitro and in vivo. This may present a novel
treatment option for decreasing TMZ toxicity and improving patient
quality of life.
Acknowledgements
The present work was supported by the National
Science Foundation of China (grant nos. 81330,016 and 31171020 to
Dr Dezhi Mu; grant nos. 81172174 and 81270724 to Dr Yi Qu), the
Major State Basic Research Development Program (grant no.
2013CB967404), grants from the Ministry of Education of China
(grant nos. 313037 and 20110181130002), the grant from the State
Commission of Science Technology of China (grant nos. 2012BAI04B04
and 2012BAI04B04), the grant from the Science and Technology Bureau
of Sichuan province (grant no. 2011JTD0005), the National Science
Foundation of China, (grant no. 81501301 to Mr. Jing Gan) and the
grant of the clinical discipline program (neonatology) from the
Ministry of Health of China (grant no. 1311200003303).
References
|
1
|
Sathornsumetee S and Rich JN: New
treatment strategies for malignant gliomas. Expert Rev Anticancer
Ther. 6:1087–1104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mrugala MM and Chamberlain MC: Mechanisms
of disease: Temozolomide and glioblastoma-look to the future. Nat
Clin Pract Oncol. 5:476–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Friedman HS, Kerby T and Calvert H:
Temozolomide and treatment of malignant glioma. Clin Cancer Res.
6:2585–2597. 2000.PubMed/NCBI
|
|
4
|
Osoba D, Brada M, Yung WK and Prados M:
Health-related quality of life in patients treated with
temozolomide versus procarbazine for recurrent glioblastoma
multiforme. J Clin Oncol. 18:1481–1491. 2000.PubMed/NCBI
|
|
5
|
Margison GP, Santibáñez Koref MF and Povey
AC: Mechanisms of carcinogenicity/chemotherapy by O6-methylguanine.
Mutagenesis. 17:483–487. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hirose Y, Berger MS and Pieper RO: p53
effects both the duration of G2/M arrest and the fate of
temozolomide-treated human glioblastoma cells. Cancer Res.
61:1957–1963. 2001.PubMed/NCBI
|
|
7
|
Quiros S, Roos WP and Kaina B: Processing
of O6-methylguanine into DNA double-strand breaks requires two
rounds of replication whereas apoptosis is also induced in
subsequent cell cycles. Cell Cycle. 9:168–178. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ahluwalia MS, Xie H, Dahiya S,
Hashemi-Sadraei N, Schiff D, Fisher PG, Chamberlain MC, Pannullo S,
Newton HB, Brewer C, et al: Efficacy and patient-reported outcomes
with dose-intense temozolomide in patients with newly diagnosed
pure and mixed anaplastic oligodendroglioma: A phase II multicenter
study. J Neurooncol. 122:111–119. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Taphoorn MJ, Henriksson R, Bottomley A,
Cloughesy T, Wick W, Mason WP, Saran F, Nishikawa R, Hilton M,
Theodore-Oklota C, et al: Health-Related quality of life in a
randomized phase III study of bevacizumab, temozolomide, and
radiotherapy in newly diagnosed glioblastoma. J Clin Oncol.
33:2166–2175. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marumoto T, Honda S, Hara T, Nitta M,
Hirota T, Kohmura E and Saya H: Aurora-A kinase maintains the
fidelity of early and late mitotic events in HeLa cells. J Biol
Chem. 278:51786–51795. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seki A, Coppinger JA, Jang CY, Yates JR
and Fang G: Bora and the kinase Aurora a cooperatively activate the
kinase Plk1 and control mitotic entry. Science. 320:1655–1658.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW,
Sahin A, Brinkley BR and Sen S: Tumour amplified kinase STK15/BTAK
induces centrosome amplification, aneuploidy and transformation.
Nature Genet. 20:189–193. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sen S, Zhou H and White RA: A putative
serine/threonine kinase encoding gene BTAK on chromosome 20q13 is
amplified and overexpressed in human breast cancer cell lines.
Oncogene. 14:2195–2200. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Van Brocklyn JR, Wojton J, Meisen WH,
Kellough DA, Ecsedy JA, Kaur B and Lehman NL: Aurora-A inhibition
offers a novel therapy effective against intracranial glioblastoma.
Cancer Res. 74:5364–5370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang H, Ou CC, Feldman RI, Nicosia SV,
Kruk PA and Cheng JQ: Aurora-A kinase regulates telomerase activity
through c-Myc in human ovarian and breast epithelial cells. Cancer
Res. 64:463–467. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Katayama H, Sasai K, Kawai H, Yuan ZM,
Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA and Sen
S: Phosphorylation by aurora kinase A induces Mdm2-mediated
destabilization and inhibition of p53. Nat Genet. 36:55–62. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qin L, Tong T, Song Y, Xue L, Fan F and
Zhan Q: Aurora-A interacts with Cyclin B1 and enhances its
stability. Cancer Lett. 275:77–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sasayama T, Marumoto T, Kunitoku N, Zhang
D, Tamaki N, Kohmura E, Saya H and Hirota T: Over-expression of
Aurora-A targets cytoplasmic polyadenylation element binding
protein and promotes mRNA polyadenylation of Cdk1 and cyclin B1.
Genes Cells. 10:627–638. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang S, He S, Zhou X, Liu M, Zhu H, Wang
Y, Zhang W, Yan S, Quan L, Bai J and Xu N: Suppression of Aurora-A
oncogenic potential by c-Myc downregulation. Exp Mol Med.
42:759–767. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carol H, Boehm I, Reynolds CP, Kang MH,
Maris JM, Morton CL, Gorlick R, Kolb EA, Keir ST, Wu J, et al:
Efficacy and pharmacokinetic/pharmacodynamic evaluation of the
Aurora kinase A inhibitor MLN8237 against preclinical models of
pediatric cancer. Cancer Chemother Pharmacol. 68:1291–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huck JJ, Zhang M, McDonald A, Bowman D,
Hoar KM, Stringer B, Ecsedy J, Manfredi MG and Hyer ML: MLN8054, an
inhibitor of Aurora A kinase, induces senescence in human tumor
cells both in vitro and in vivo. Mol Cancer Res. 8:373–384. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hata T, Furukawa T, Sunamura M, Egawa S,
Motoi F, Ohmura N, Marumoto T, Saya H and Horii A: RNA interference
targeting aurora kinase a suppresses tumor growth and enhances the
taxane chemosensitivity in human pancreatic cancer cells. Cancer
Res. 65:2899–2905. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dai L, Cui X, Zhang X, Cheng L, Liu Y,
Yang Y, Fan P, Wang Q, Lin Y, Zhang J, et al: SARI inhibits
angiogenesis and tumour growth of human colon cancer through
directly targeting ceruloplasmin. Nat Commun. 7:19962016.
View Article : Google Scholar
|
|
24
|
Dai L, Cheng L, Zhang X, Jiang Q, Zhang S,
Wang S, Li Y, Chen X, Du T, Yang Y, et al: Plasmid-based
STAT3-siRNA efficiently inhibits breast tumor growth and metastasis
in mice. Neoplasma. 58:538–547. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang W, Dai LX, Zhang S, Yang Y, Yan N,
Fan P, Dai L, Tian HW, Cheng L, Zhang XM, et al: Regulation of
epidermal growth factor receptor signaling by plasmid-based
microRNA-7 inhibits human malignant gliomas growth and metastasis
in vivo. Neoplasma. 60:274–283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cammareri P, Scopelliti A, Todaro M,
Eterno V, Francescangeli F, Moyer MP, Agrusa A, Dieli F, Zeuner A
and Stassi G: Aurora-a is essential for the tumorigenic capacity
and chemoresistance of colorectal cancer stem cells. Cancer Res.
70:4655–4665. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hegi ME, Liu L, Herman JG, Stupp R, Wick
W, Weller M, Mehta MP and Gilbert MR: Correlation of
O6-methylguanine methyltransferase (MGMT) promoter methylation with
clinical outcomes in glioblastoma and clinical strategies to
modulate MGMT activity. J Clin Oncol. 26:4189–4199. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang Z, Pore N, Cerniglia GJ, Mick R,
Georgescu MM, Bernhard EJ, Hahn SM, Gupta AK and Maity A:
Phosphatase and tensin homologue deficiency in glioblastoma confers
resistance to radiation and temozolomide that is reversed by the
protease inhibitor nelfinavir. Cancer Res. 67:4467–4473. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Woo SR, Ham Y, Kang W, Yang H, Kim S, Jin
J, Joo KM and Nam DH: KML001, a telomere-targeting drug, sensitizes
glioblastoma cells to temozolomide chemotherapy and radiotherapy
through DNA damage and apoptosis. Biomed Res Int. 2014:7474152014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Goellner EM, Grimme B, Brown AR, Lin YC,
Wang XH, Sugrue KF, Mitchell L, Trivedi RN, Tang JB and Sobol RW:
Overcoming temozolomide resistance in glioblastoma via dual
inhibition of NAD+ biosynthesis and base excision repair. Cancer
Res. 71:2308–2317. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park I, Mukherjee J, Ito M, Chaumeil MM,
Jalbert LE, Gaensler K, Ronen SM, Nelson SJ and Pieper RO: Changes
in pyruvate metabolism detected by magnetic resonance imaging are
linked to DNA damage and serve as a sensor of temozolomide response
in glioblastoma cells. Cancer Res. 74:7115–7124. 2014. View Article : Google Scholar : PubMed/NCBI
|