Introduction
Congenital leukemia is a rare disease, with a
reported incidence of one to five cases per million births
(1). It represents the second most
common cancer in infants (while the first is neuroblastma) in
newborns and typically has a poor prognosis (2). Transient abnormal myelopoiesis (TAM),
also called transient myeloproliferative disorder, is a
pre-leukemia disorder that occurs in Down syndrome (DS) and non-DS
individuals. Morphologically, cytochemically and clinically, the
presentation of TAM may be indistinguishable from acute
megakaryoblastic leukemia (AMKL; commonly French-American-British
type M7) (2). AMKL has a good
prognosis, as it has been observed to enter spontaneous remission
in ~80% of cases (3). TAM is
associated with trisomy 21 in blast cells and mutations of the GATA
binding protein 1 (GATA1) gene (4), which synthesizes a growth factor
essential for the differentiation of erythroid and megakaryocytic
cells. This disorder must be continually monitored, as it has been
observed to develop into AMKL in 16–30% of cases during the first
four years of life (5). The
differentiation between TAM and AMKL is primarily based on the
GATA1 mutations, moreover the latter diverges from the
former in morphological characteristics of megakaryoblasts and
positive cluster of differentiation, typically CD41/42 or CD 6 on
flow cytometry (6). Furthermore, AMKL
blast cells exhibit an additional cytogenetic abnormalities,
including extra copies of chromosome 8 (7). TAM has been identified in three
contexts: Patients with DS, patients with trisomy 21 mosaicism and
patients without DS. TAM occurs in ~10% of DS cases (7) and 7–16% of trisomy 21 mosaicism cases
(3,8,9); however,
the incidence of TAM in patients without DS is extremely rare and
only 16 cases have been reported in the literature (6,10). The
first case of trisomy 21-associated TAM was described erroneously
as a leukemoid reaction in 1980 by Sikand et al (11). The present report describes the case
of a newborn patient who exhibited clinical manifestations of acute
leukemia five days after birth, in addition to a normal karyotype,
the presence of trisomy 21 only in blast cells and spontaneous
remission.
Case report
Presentation
The newborn female patient (gestational age, 37
weeks; weight, 2,570 g; length, 46.3 cm), who was a quadruplet
delivered by Caesarean section, was transferred to the neonatal
intensive care unit of San Carlo Hospital (Potenza, Italy) on May
2013 two days after birth, due to a pale appearance, moderate
hypotonia, hepatomegaly and hypertransaminasemia. The parents were
non-consanguineous, young and healthy, with no reported familial
history of DS. All pregnancy infections were excluded, and vaginal
and rectal swabs were negative for bacteria colonization. The
initial clinical examination yielded the following results: Red
blood cell count, 2,860,000/µl (4,800,000–7,200,000/µl); white
blood cell count, 134,000/µl (1,500–10,000/µl); platelet count,
253,000/µl (259,000–615,000/µl); hemoglobin level, 12.10 g/dl
(12,7–18,3 g/dl); hematocrit level, 36.8% (52%); prothrombin time,
36 sec (10.8–13); partial thromboplastin time, 44.9 sec (26.2–36);
international normalized ratio, 1.95 (1.01); fibrinogen, 213.0
mg/dl (207–321 mg/dl); antithrombin III, 81% (60–90%); C-reactive
protein level, 20 mg/dl (<0.5 mg/dl); neutrophil granulocytes,
45.5% (34%); lymphocytes, 51.3% (40%); eosinophil granulocytes,
0.1% (3.1%); and basophil granulocytes, 0.2% (0.4%). The levels of
aspartate transaminase (1,011 UI/dl; normal values 15–131 UI/l),
alanine transaminase (515 UI/dl; normal values 28–300 UI/l) and
lactate dehydrogenase (9,957 UI/dl; 150–360 UI/l) were elevated.
The patient had normal ammonia levels, and other metabolic
examinations were within the normal ranges. Chest X ray was
negative. Blood and urine cultures were collected and the patient
commenced a course of antibiotic therapy for seven days, due to
suspicion of infection (ampicillin 50 mg/kg every 12 h and
gentamicin, 4 mg/kg every 24 h). Fresh plasma was administered to
correct coagulation. Following two days of treatment, the platelet
count began to decrease (fourth day of life, 93,000/µl; fifth day
of life, 73,000/µl). Standard cytogenetic and flow cytometric
analyses were performed on peripheral blood and bone marrow
samples. The results indicated 65% blast cells, a feature
compatible with acute leukemia (immunophenotype characteristics
between M0 vs. M7) (12). Karyotype
analysis of the of bone marrow and peripheral blood cells revealed
trisomy 21 (46, XX+21) in all blast cells.
Six days following birth, the infant was referred to
Bambino Gesù Children's Hospital (Rome, Italy). Upon physical
examination, the patient exhibited no visible DS characteristics.
Persistent hepatomegaly and occasional petechiae were observed. The
results of the neurological examination were normal for the
gestational age and the bregmatic fontanelle was normotensive and
normally pulsating. The patient exhibited normal vital signs and
was afebrile. An echocardiogram identified a patent foramen ovale.
Ultrasonography of the brain, abdomen and kidneys was normal. Bone
marrow cells immunophenotyping demonstrated that ~31% of cells were
CD33 heterogeneous, CD117-positive, CD34 heterogeneous, CD71 dull,
CD45 dull and DR negative. A single-base deletion (c.150delG) in
exon 2 of the GATA1 gene was identified in peripheral blood
cell samples, after the admission to our center. The patient also
exhibited a reduction in the platelet count, without any specific
treatment, to a 40,000/µl nadir. From one month and five days, the
platelet count was observed to increase again. Prior to discharge,
cultures of fibroblasts and of the Epstein-Barr virus
lymphoblastoid T-cell line-reactive test were negative. Considering
these clinical features and the presence of a GATA1
mutation, which could indicate transient leukemia, it was decided
that chemotherapy would not be administered. The patient
subsequently demonstrated a healthy clinical condition with normal
values for the aforementioned clinical features. A bone marrow
aspiration, conducted at the seven-month follow-up, revealed a
normal karyotype and the absence of leukemic cells. Simultaneously,
a bone marrow biopsy confirmed the absence of leukemic cell
infiltration.
Written informed consent was obtained from the
patient's family for the publication of the current case report and
the accompanying images.
Flow cytometry
Flow cytometry was performed on blood or bone marrow
specimens using 3- or 4-color antibody panels against a variety of
lymphoid, myelomonocytic, and megakaryocytic antigens. The
antibodies (and clones) used included (all antibodies from
Pharmingen; BD Biosciences, San Jose, CA, USA, unless otherwise
stated): anti-CD1a (HI149), CD3 (SK7), CD4 (SK3), CD5 (L17F12), CD7
(4H9), CD8 (SK1), CD10 (W8E7), CD11b (D12), CD13 (L138), CD14
(M/P9), CD15 (MMA), CD16 (NKP15), CD19 (SJ25C1), CD20 (L27), CD22
(S-HCL-1), CD33 (P67.6), CD34 (8G12), CD36 (FA6.152; Beckman
Coulter, Inc., Miami, FL, USA), CD38 (HB7), CD41 (P2; Beckman
Coulter), CD45 (2D1), CD56 (MY31), CD61 (RUUPL7F12), CD64 (10;
Ancell, Bayport, MN), CD71 (L01.1/M-A712), CD117 (2B8), HLA-DR
(L243), surface light chains kappa (TB28-2) and lambda (1-155-2),
glycophorin A (GA-R2) and appropriate isotypic control antibodies
(X40, X39). Specimen processing and antibody staining were
performed as previously described (13). All data were acquired using 3-color
FACSort or 4-color FACSCalibur flow cytometers with Cellquest
software (BD Biosciences). The data was analyzed by cluster
analysis with Paint-a-Gate Software (BD Biosciences).
Cytogenetics studies
Firstly, during hospitalization, chromosomal
analyses of unstimulated bone marrow of patient were conducted on
20 metaphases revealing an abnormal karyotype: 47, XX, +21
(Fig. 1B) Subsequently, the
chromosomal analyses were extended to two other issues: Peripheral
blood cells and dermal fibroblasts using a G-banding technique, as
previously described (12,14). A total of 20 metaphases for dermal
fibroblasts and 50 metaphases for peripheral blood cells, were
analyzed demonstrating a normal karyotype: 46, XX (Fig. 1A), in both cases. Structural and
numerical anomalies were recorded and karyotyped according to the
International System for Human Cytogenetic Nomenclature (2013) or
all three samples (14).
In order to investigate this result, fluorescence
in situ hybridization (FISH) was performed using a probe
specific for the 21q22.2 region. FISH was conducted as previously
described (14), using a Vysis LSI 21
SpectrumOrange Probe Kit (locus 21q22.13-q22.2; Abbott Molecular,
Illinois, USA) to identify the critical region of DS. Analysis of
500 nuclei from bone marrow samples identified the presence of
three signals in 70% of the cells (Fig.
2A); this result represents an abnormal condition. Instead, the
remaining 30% of the cells of bone marrow exhibited two signals, as
expected in normal conditions. Conversely, analysis of the
peripheral blood cells and dermal fibroblasts (500 nuclei) revealed
only two signals per cell (Fig. 2B and
C).
All nuclei were analyzed using a Nikon Eclipse E1000
epifluorescence microscope, equipped with a CoolSNAP fx
charge-coupled device camera (Photometrics, Tucson, AZ, USA). Image
capturing and processing were performed using Genikon software
v3.6.16 (Nikon Corporation, Tokyo, Japan).
Molecular studies
Amplification of GATA1 exon 2 genomic DNA
(genomic DNA accession no. AF196971; complementary DNA accession
no. NM_002049) was performed in 20 µl containing 10 µl KAPA 2G Fast
HS ReadyMix PCR Kit (Kapa Biosystems, Inc., Wilmington, MA, USA), 1
uM primers (FW-5′-TCTGTCCTCGCAGGTTAATCCR-3′,
RV-5′-TATTCTGACCTAGCCAAGGATCTC-3′) and 2 µl of DNA (50 ng).
The cycling profile was set at 96°C initial
denaturation for 3 min, followed by 35 cycles of 10 sec at 96°C, 10
sec at 58°C, 2 sec at 72°C, and a final extension at 72°C for 5 min
in a GeneAmp® PCR system 2700 (Applied Biosystems;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). PCR products
were enzymatically purified using Illustra ExoProStar reagent
containing phosphatase and exonuclease (GE Healthcare Life
Sciences, Chalfont, UK). Sequencing reactions were carried out
using Big Dye® Terminator v3.1 Cycle Sequencing Kit
(Applied Biosystems, Foster City, CA, USA) according to the
manufacturer's protocol. The reaction products were purified after
sequencing by using DyeEx® 2.0 Spin Kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Direct sequencing was
performed in both directions for all samples on an automated
sequencer ABI Prism® 3130 Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). GATA1 sequencing
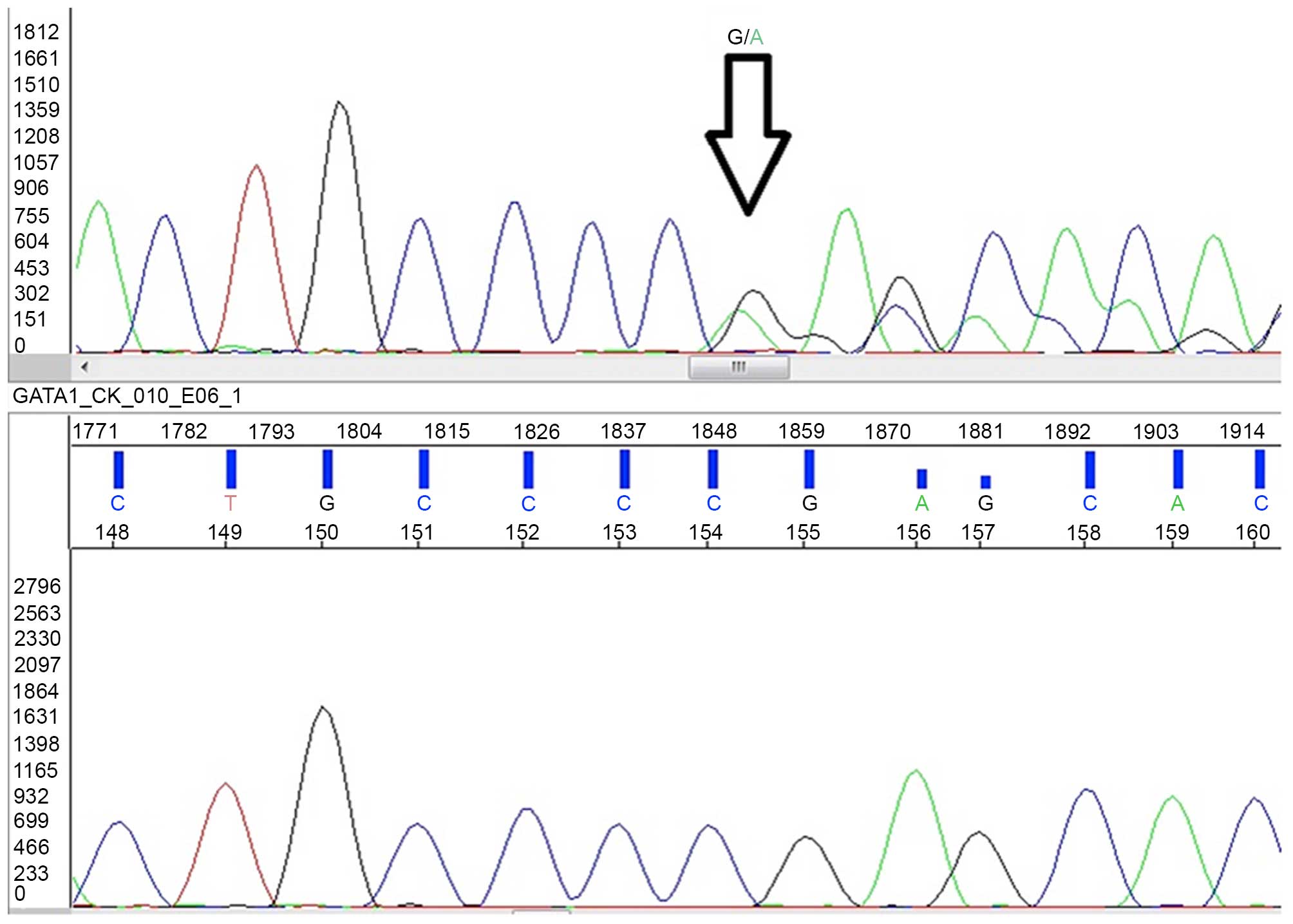
identified the deletion of a single base (c.150delG) in exon 2,
that caused a frameshift resulted in premature sequence termination
(p.Ser51Alafs*86; Fig. 3) (15).
Discussion
To the best of our knowledge, this is the first
Italian case of TAM in a non-DS infant with trisomy 21 only in
blast cells; this condition differs from congenital leukemia due to
its spontaneous remission (2). In the
present case, considering the improvement of clinical features,
coagulation and inflammatory markers, in addition to the gradual
increase in the number of platelets, it was assumed that acute
leukemia was a transitory clinical episode and that the blasts had
been progressively reduced by apoptosis.
Differentiating between TAM and congenital acute
leukemia is challenging and it is important to reach a rapid
diagnosis. The initial approach is to exclude the possibility of
leukemoid reactions due to other conditions, including infection,
hypoxia and hemolytic disease (1). It
is crucial to perform a complete blood cell count with a peripheral
blood smear and a bone marrow aspiration with immunophenotyping and
cytogenetic testing (1). The typical
presentation of transient leukemia in infants is leukocytosis,
often with a higher percentage of blasts in the peripheral blood
compared with in the bone marrow (16). Hepatosplenomegaly, pericardial or
pleural effusions, and liver disease are common features. The
blasts are frequently of the megakaryocytic or erythroid lineages
(16). In the event that a newborn
with a normal karyotype and trisomy 21 only in blasts presents
symptoms typical of congenital leukemia, the best approach is to
‘wait and see’ and to perform GATA1 gene sequencing as soon
as possible. If the patient's condition deteriorates, it may be
advisable to commence low-dose chemotherapy (17). The presence of a GATA1 mutation
can confirm a diagnosis of TAM and therefore determine the
non-therapeutic approach.
The GATA1 gene is located on the short (p)
arm of the X chromosome at position 11.23; the encoded protein is a
transcription factor involved in the differentiation of
megakaryocytic precursors of erythrocytes (6,10). The
mutations are somatic, not inherited, and occur during fetal
development; the increased risk of leukemia applies only to
patients with an extra copy of chromosome 21 (18). GATA1 may be involved in folate
metabolism and oxidative stress in trisomy cells, modifying the
repair mechanisms of DNA damage and creating pre-leukemic clones
with a selective growth advantage (19). Environmental, immune and reproductive
factors may contribute to the mutation process (18). The mutations on exon 2 of the
GATA1 transcription factor gene have been revealed to
exhibit 100% penetrance in TAM (19).
A previous study analyzed the mutation spectrum of sequence
alterations at GATA1 exon 2 in DS TAM/AMKL cases, revealing
that the mutations may be insertions/deletions/duplications or base
substitutions (19). The mutation
detected in this case was a deletion of a single base (c.150delG),
which determines an accumulation of uracil in cells under oxidative
stress. Moreover, substitutions of a single base in the
GATA1 gene have been reported in 6 cases of TAM; in a
retrospective series, a myelodysplastic syndrome or acute myeloid
leukemia (20), often of the acute
megakaryoblastic type, developed in ~30% of patients with TAM
(16).
Once a diagnosis of TAM has been determined, it is
necessary to follow-up with the patients to detect the possible
occurrence of leukemia. In a previous study, out of 16 cases of TAM
in non-DS patients, 5 developed subsequent leukemia, with 3
developing AMKL and 2 developing non-AMKL acute myeloid leukemia
(10), and a total of 9 patients
subsequently required chemotherapy (21).
The present case study adds another example of
spontaneous remission of TAM to the literature, contributing to the
discovery of an additional mutation of GATA1 gene. Furthermore, the
findings of the current study may aid clinicians in decision-making
when treating cases of TAM. Firstly, the best way of approaching a
suspected case of TAM is waiting without starting chemotherapy.
Secondly, if a case is strongly suspected to be TAM, performing
GATA1 gene sequencing may confirm the diagnostic hypothesis.
Further investigations are required to explain how blast cells
disappear spontaneously, as if it obtained a selective
disadvantage.
References
|
1
|
Orbach D, Sarnacki S, Brisse HJ,
Gauthier-Villars M, Jarreau PH, Tsatsaris V, Baruchel A, Zerah M,
Seigneur E, Peuchmaur M and Doz F: Neonatal cancer. Lancet Oncol.
14:e609–e620. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Isaacs H Jr: Fetal and neonatal leukemia.
J Pediatr Hematol Oncol. 25:348–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gamis AS, Alonzo TA, Gerbing RB, Hilden
JM, Sorrell AD, Sharma M, Loew TW, Arceci RJ, Barnard D, Doyle J,
et al: Natural history of transient myeloproliferative disorder
clinically diagnosed in Down syndrome neonates: A report from the
children's oncology group study A2971. Blood. 118:6752–6759; quiz
6996. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vyas P and Crispino JD: Molecular insights
into Down syndrome-associated leukemia. Curr Opin Pediatr. 19:9–14.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bull MJ: Committee on Genetics: Health
supervision for children with Down syndrome. Pediatrics.
128:393–406. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsai MH, Hou JW, Yang CP, Yang PH, Chu SM,
Hsu JF, Chiang MC and Huang HR: Transient myeloproliferative
disorder and GATA1 mutation in neonates with and without Down
syndrome. Indian J Pediatr. 78:826–832. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schifferli A, Hitzler J, Bartholdi D,
Heinimann K, Hoeller S, Diesch T and Kühne T: Transient
myeloproliferative disorder in neonates without Down syndrome: Case
report and review. Eur J Haematol. 94:456–462. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Massey GV, Zipursky A, Chang MN, Doyle JJ,
Nasim S, Taub JW, Ravindranath Y, Dahl G and Weinstein HJ:
Children's Oncology Group (COG): A prospective study of the natural
history of transient leukemia (TL) in neonates with Down syndrome
(DS): children's oncology group (COG) study POG-9481. Blood.
107:4606–4613. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klusmann JH, Creutzig U, Zimmermann M,
Dworzak M, Jorch N, Langebrake C, Pekrun A, Macakova-Reinhardt K
and Reinhardt D: Treatment and prognostic impact of transient
leukemia in neonates with Down syndrome. Blood. 111:2991–2998.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Apollonsky N, Shende A, Ouansafi I, Brody
J, Atlas M and Aygun B: Transient myeloproliferative disorder in
neonates with and without Down syndrome: A tale of 2 syndromes. J
Pediatr Hematol Oncol. 30:860–864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sikand GS, Taysi K, Strandjord SE,
Griffith R and Vietti TJ: Trisomy 21 in bone marrow cells of a
patient with a prolonged preleukemic phase. Med Pediatr Oncol.
8:237–242. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chisté M, Vrotsos E, Zamora C and Martinez
A: Chronic lymphocytic leukemia/small lymphocytic lymphoma
involving the aortic valve. Ann Diagn Pathol. 17:295–297. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Karandikar NJ, Aquino DB, McKenna RW and
Kroft SH: Transient myeloproliferative disorder and acute myeloid
leukemia in Down syndrome. An immunophenotypic analysis. Am J Clin
Pathol. 116:204–210. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Simons A, Shaffer LG and Hastings RJ:
Cytogenetic Nomenclature: Changes in the ISCN 2013 Compared to the
2009 Edition. Cytogenet Genome Res. 141:1–6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lum SH, Choong SS, Krishnan S, Mohamed Z
and Ariffin H: GATA1 mutations in a cohort of Malaysian children
with Down syndrome-associated myeloid disorder. Singapore Med J.
57:320–324. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wolfe LC, Weinstein HJ and Ferry JA: Case
records of the Massachusetts general hospital. Weekly
clinicopathological exercises. Case 19–200. A five-day-old girl
with leukocytosis and a worsening rash from birth. N Engl J Med.
348:2557–2566. 2003.PubMed/NCBI
|
|
17
|
Ohkawa T, Miyamoto S, Sugie M, Tomizawa D,
Imai K, Nagasawa M, Morio T, Mizutani S and Takagi M: Transient
abnormal myelopoiesis in non-Down syndrome neonate. Pediatr Int.
57:e14–e17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mezei G, Sudan M, Izraeli S and Kheifets
L: Epidemiology of childhood leukemia in the presence and absence
of Down syndrome. Cancer Epidemiol. 38:479–489. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cabelof DC, Patel HV, Chen Q, van Remmen
H, Matherly LH, Ge Y and Taub JW: Mutational spectrum at GATA1
provides insights into mutagenesis and leukemogenesis in Down
syndrome. Blood. 114:2753–2763. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Homans AC, Verissimo AM and Vlacha V:
Transient abnormal myelopoiesis of infancy associated with trisomy
21. Am J Pediatr Hematol Oncol. 15:392–399. 1993.PubMed/NCBI
|
|
21
|
Ono R, Hasegawa D, Hirabayashi S, Kamiya
T, Yoshida K, Yonekawa S, Ogawa C, Hosoya R, Toki T, Terui K, et
al: Acute megakaryoblastic leukemia with acquired trisomy 21 and
GATA1 mutations in phenotypically normal children. Eur J Pediatr.
174:525–531. 2015. View Article : Google Scholar : PubMed/NCBI
|