Introduction
Nasopharyngeal carcinoma (NPC) is a malignant
disease characterized by its unique geographic distribution, with
Southern China and Southeast Asia holding the highest incidence
rates, with an annual incidence of 15–50 cases per 100,000 people
(1–4).
The main etiological factors of NPC are genetic susceptibility,
environment factors and Epstein-Barr virus infection (1). Radiotherapy is the recommended treatment
for non-metastatic NPC at early stage due to its complex anatomic
location and high radiosensitivity (5). Combined chemoradiotherapy is more
effective in treating NPC patients with locally advanced stages or
lymph node metastasis, which account for the majority of NPC types
(3,4).
Although the treatment results of NPC have improved in the past
years, its overall survival (OS) rates at 5 years are still
unsatisfactory (3). Thus, it is
necessary and helpful to explore new agents that could achieve
better therapeutic efficacy of NPC than the current ones.
Sodium butyrate (NaBu) is the sodium salt of butyric
acid, one of the short chain fatty acids, which is naturally
produced by symbiotic bacteria in the gastrointestinal tract
through fermenting dietary fibers (6–8). Histone
deacetylases (HDACs) are key epigenetic regulators that can
regulate the gene expression profile by directly interacting with
other proteins such as transcription factors (6). Their abnormal overexpression has been
confirmed to contribute to carcinogenesis in multiple cancers
(6). Therefore, HDAC inhibitors
(HDACis) serve as a promising new class of anti-cancer tumor
agents, and various agents of this type have been approved
(6,8).
NaBu, as a potent HDACi, has a great potential in cancer treatment
and prevention (9). Its anti-tumor
abilities, including proliferation inhibition and apoptosis
induction (9), have been validated in
several cancers, including colorectal cancer, prostate cancer,
breast cancer and lung cancer (9–11).
However, NaBu's influence on NPC cells has been rarely studied.
Ca2+ is a ubiquitous second messenger in
signaling transduction, and the fluctuation of intracellular
Ca2+ levels is involved in the regulation of multiple
physiological functions, including cell growth, survival, apoptosis
and migration (12). In non-excitable
cells, store-operated Ca2+ entry (SOCE) is an important
pathway for Ca2+ influx to refill the intracellular
Ca2+ stores, mainly located in the endoplasmic reticulum
(ER), which is mediated by the Ca2+ release-activated
Ca2+ (CRAC) channel (13,14).
Calcium release-activated calcium channel protein 1 (Orai1) and
stromal interaction molecule 1 (Stim1) are two essential components
of the CRAC channel (13,14). Upon activation, following
oligomerization and translocation, Stim1, a single-pass
transmembrane protein located in the membrane of ER, recruits
Orai1, a cytomembrane protein, to form the functional CRAC channel,
which stimulates Ca2+ influx (13,14). Under
certain circumstances, including endogenous mutations and exogenous
drugs stimulation, the Orai1/Stim1 CRAC channel may overexcite, and
the aberrant overloaded intracellular Ca2+ concentration
would cause a series of pathological responses such as apoptosis
(15,16). Interrupting the SOCE process by
knocking down Orai1/Stim1 or blocking it with specific inhibitors
could counteract the apoptosis of cells induced by anti-tumor
agents such as cisplatin, gypenosides and oxaliplatin in several
cancers (7,15,16). SOCE
was also confirmed to be necessary for the apoptosis induced by
NaBu in colon cancer cells (17).
Thus, in the present study, various experiments were
performed to test the anti-tumor efficacy of NaBu in NPC cells, and
the roles of SOCE in NaBu-induced apoptosis in NPC cells were
explored.
Materials and methods
Cell culture and transfection
The NPC cell lines 5–8F and 6–10B were obtained from
the Cancer Research Institute of Sun Yat-sen University (Guangzhou,
China). The cells were cultured in RPMI-1640 medium (Sigma-Aldrich;
Merck Millipore, Darmstadt, Germany) supplemented with 10% fetal
calf serum (Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) at 37°C in a humidified 5% CO2 atmosphere.
5-8F and 6–10B cells were transfected with small
interefering RNA (siRNA) against Orai1 and Stim1 using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Cells were collected for
downstream analyses 48 h later,. The sequences of the siRNAs were
listed elsewhere (18).
MTT assay
MTT (Sigma-Aldrich; Merck Millipore) assay was
performed to evaluate the proliferation ability of 5–8F and 6–10B
cells treated with NaBu. Cells were seeded at a density of 2,000
cells/well in 96-well plates with 200 µl culture medium containing
NaBu at different concentrations. Then, the cells were
consecutively cultured for 72 h. Every 24 h, 20 µl 5 mg/ml MTT
solution was added into the corresponding well, and the cells were
cultured for another 4 h. Then, the solution was replaced with 150
µl dimethylsulfoxide (Sigma-Aldrich; Merck Millipore), followed by
gentle agitation of the plates for 15 min at room temperature.
Finally, the absorbance at 492 nm was measured to represent the
cell viability.
Colony formation assay
Colony formation assay was performed as described
earlier (19). First, 5–8F and 6–10B
cells were treated with 5 mM NaBu for 48 h. Then, the treated cells
were trypsinized and seeded in 6-well plates at a density of 1,000
cells/well. Untreated 5–8F and 6–10B cells were also seeded in
6-well plates similarly. Eight days later, the cultured cells were
fixed with paraformaldehyde and stained with crystal violet.
Colonies containing ≥50 cells were counted under an inverse
microscope (Nikon Corporation, Tokyo, Japan), and the colony
formation ability was subsequently analyzed.
Apoptotic analysis by flow
cytometry
Apoptotic analysis by flow cytometry was conducted
using the Annexin V-FITC Apoptosis Detection kit (Vazyme,
Piscataway, NJ, USA) according to the manufacturer's protocol.
Briefly, 2×106 cells, both treated and untreated with
NaBu, were collected and washed by pre-cooling PBS twice. Then,
cells were resuspended in staining buffer and stained with 5 µl
fluorescein isothiocyanate and 5 µl propidium iodide for 10 min in
the dark at room temperature. Finally, the cell samples were
analyzed by FACSCalibur (BD Biosciences, San Jose, CA, USA).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA extraction and RT-qPCR were performed according
to a previous procedure (2). Briefly,
total RNA was extracted using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) and retrotranscribed into transcripts with
the HiScript® II Q RT SuperMix for qPCR kit (Vazyme).
Then, qPCR was conducted on a MJ Mini™ thermal cycler (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) using AceQ® qPCR
SYBR® Green Master Mix (Vazyme). The qPCR cycle program
was as follows: 95°C for 5 min, followed by 40 cycles at 94°C for
10 sec and 60°C for 30 sec. The relative messenger RNA expression
levels of Orai1 and Stim1 were calculated using the
2−ΔΔCq method (20)
following normalized to GAPDH. The primers for Orai1, Stim1 and
GAPDH are listed in Table I.
 | Table I.Primer sequences for quantitative
polymerase chain reaction. |
Table I.
Primer sequences for quantitative
polymerase chain reaction.
| Gene names | Primer
sequences | Product sizes |
|---|
| Orai1 | Forward:
5′-GACTGGATCGGCCAGAGTTAC-3′ | 116 bp |
|
| Reverse:
5′-GTCCGGCTGGAGGCTTTAAG-3′ |
|
| Stim1 | Forward:
5′-AGTCACAGTGAGAAGGCGAC-3′ | 79
bp |
|
| Reverse:
5′-CAATTCGGCAAAACTCTGCTG-3′ |
|
| GAPDH | Forward:
5′-CCAGCAAGAGCACAAGAGGAA-3′ | 157 bp |
|
| Reverse:
5′-ATGGTACATGACAAGGTGCGG-3′ |
|
Antibodies and western blotting
Rabbit anti-B-cell lymphoma (Bcl)-xL (cat. no.
10783-1-AP), anti-Bcl-2 (cat. no. 12789-1-AP), anti-caspase 3
(Casp3) (cat. no. 19677-1-AP), anti-Bcl-2-associated X protein
(Bax) (cat. no. 23931-1-AP), anti-Orai1 (cat. no. 13130-1-AP),
anti-Stim1 (cat. no. 11565-1-AP), anti-myeloid leukemia cell
differentiation protein-1 (Mcl1) (cat. no. 16225-1-AP) and
anti-survivin (cat. no. 10508-1-AP) polyclonal antibodies, and
mouse anti-β-actin monoclonal antibody (cat. no. 66009-1-Ig), were
all purchased from ProteinTech Group, Inc. (Chicago, IL, USA).
Rabbit anti-cleaved (c)-Casp3 p17 (cat. no. D175) and c-poly ADP
ribose polymerase (c-PARP) (cat. no. YC0101) polyclonal antibodies
were purchased from ImmunoWay Biotechnology Company (Plano, TX,
USA). Horseradish peroxidase (HRP)-conjugated anti-rabbit
immunoglobulin G secondary antibody was purchased from
Sigma-Aldrich (Merck Millipore).
Cells were disrupted with radioimmunoprecipitation
assay buffer (Vazyme), and the supernatants containing total
proteins were isolated by high-speed centrifugation at 4°C and
13,000 × g for 20 min. Then, the protein concentration was
determined using the Pierce BCA Protein Assay kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Following separation by 12% SDS-PAGE (40 µg/lane), the proteins
were electrophoretically transferred to polyvinylidene difluoride
membranes using a wet transfer system (Bio-Rad Laboratories, Inc.).
Subsequently, the membranes were subjected to blocking with milk,
incubation with the primary antibodies (all 1:200 dilution) at 4°C
overnight, and incubation with the secondary antibody (1:5,000
dilution) for 1 h at room temperature. Finally, the immunoreactive
bands were developed with a chemiluminescent HRP substrate (Merck
Millipore).
Intracellular Ca2+
measurements
Ca2+ measurements were conducted
according to a published study (17).
Briefly, the cells were collected and loaded with 5 µM Fluo
3-acetoxymethyl (AM) (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) for 30 min at 37°C. After washing with Hanks'
balanced salt solution (Ca2+ free) for three times, the
cells were incubated at 37°C for another 20 min to allow
de-esterification of Fluo 3-AM. Subsequently, the cells were
treated with different combinations of agents and immediately
subjected to flow cytometry analysis to detect the intracellular
Ca2+ levels. 2-Aminoethoxydiphenyl borate (APB), a SOCE
specific inhibitor, and ethylene glycol-bis(β-aminoethyl
ether)-N,N,N',N'-tetraacetic acid (EGTA), a Ca2+
chelator, were both purchased from Sigma-Aldrich (Merck
Millipore).
Statistical analyses
Statistical analyses were conducted with SPSS 18.0
statistical software (SPSS, Inc., Chicago, IL, USA) using the
Student's t-test, and all the experiments were independently
performed in triplicate. P<0.05 was considered to indicate a
statistically significant difference.
Results
NaBu induces morphological changes and
inhibits the proliferation of NPC cell lines
It has been demonstrated that NaBu can induce cell
morphological changes in multiple cancers (21). In the present study, obvious
morphological transformations induced by NaBu were observed in both
5–8F and 6–10B cells, as demonstrated by extended pseudopodia,
enlarged and compressed cell size, and increased cellular vacuoles
(Fig. 1A). These transformations
exhibited a dose- and time-dependent pattern, since higher
concentrations and longer time exposures led to bigger changes
(Fig. 1A; dose-dependent results not
shown). NaBu inhibited the proliferation of 5–8F and 6–10B cells
remarkably, as demonstrated by the reduced number of cells and
reduced size of the cell colonies in the colony formation assay
(Fig. 1B).
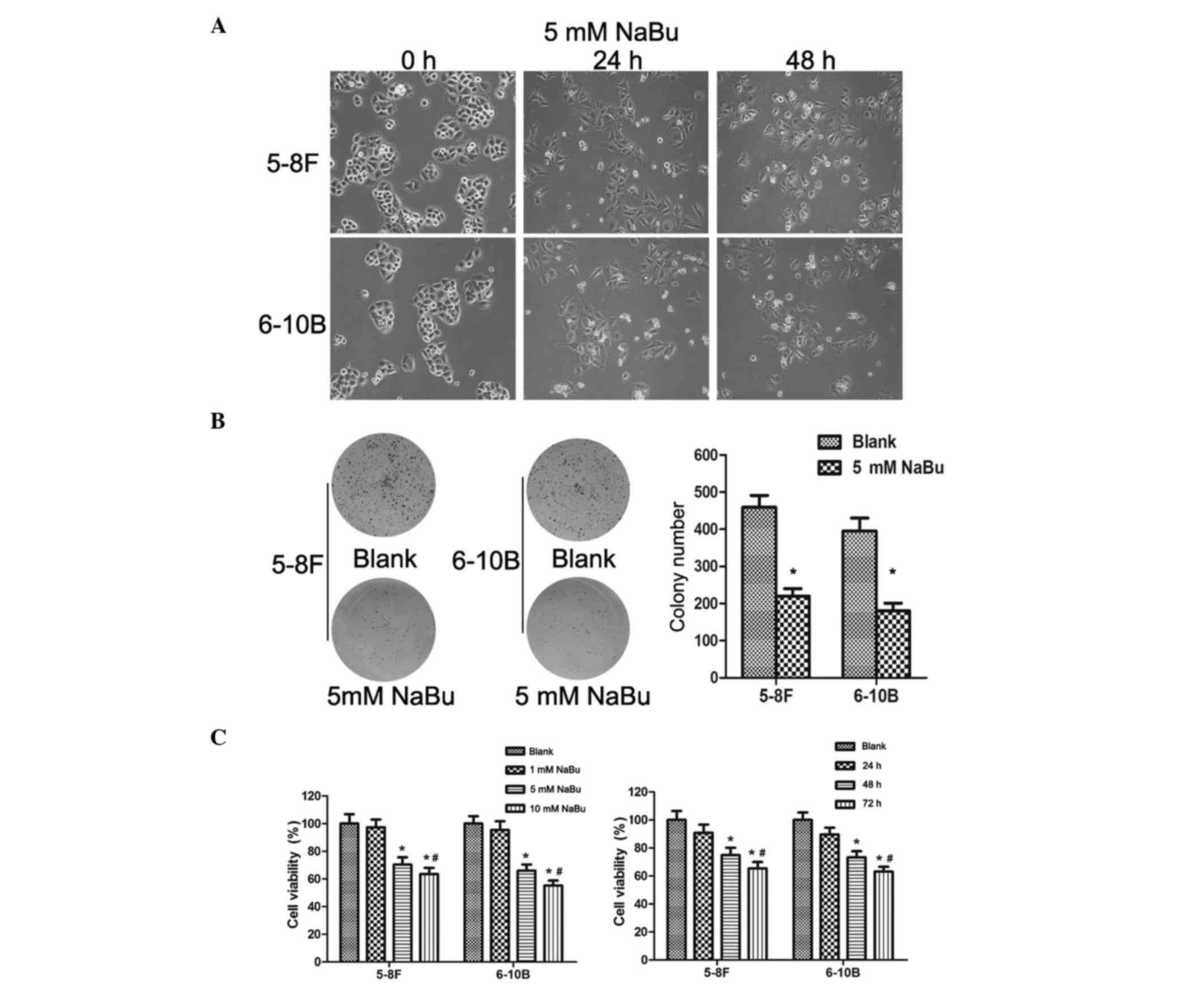 | Figure 1.NaBu induced morphological changes,
inhibited cell proliferation and impaired cell viability in NPC
cells. (A) The morphology of 5–8F and 6–10B cells treated with NaBu
changed, with extended pseudopodia, enlarged and compressed cell
size and increased cellular vacuoles, in a dose-dependent manner.
(B) NaBu inhibited the proliferation of NPC cells. The colony
number and size of 5–8F (left panel) and 6–10B (right panel) cells
were obviously decreased upon treatment with 5 mM NaBu. *P<0.05.
(C) Left panel: NaBu impaired the viability of 5–8F and 6–10B cells
in a dose-dependent manner. *P<0.05, 5 and 10 mM groups compared
with the blank and 1 mM groups. #P<0.05, 5 mM group
compared with the 10 mM group. Right panel: NaBu impaired the
viability of 5–8F and 6–10B cells in a time-dependent manner.
*P<0.05, 48 and 72 h groups compared with the blank and 24 h
groups. #P<0.05, 48 h group compared with the 72 h
group. NaBu, sodium butyrate; NPC, nasopharyngeal carcinoma. |
NaBu decreases the cell viability of
NPC cell lines in vitro
The cytotoxicity of NaBu has been confirmed in
multiple cancer types (11,22,23). The
present study explored the NaBu's cytotoxicity in NPC cells by MTT
assay. 5–8F and 6–10B cells were treated with NaBu at different
concentrations and exposure times. Under low concentrations (1 mM)
and short exposure times (24 h), there were no significant
differences in cell viability between the blank and the
NaBu-treated group (Fig. 1C).
However, the cytotoxicity of NaBu towards 5–8F and 6–10B cells
increased with higher concentrations (5 and 10 mM) and longer
exposure times (48 and 72 h) (Fig.
1C). Therefore, NaBu was cytotoxic to NPC cells, inducing a
dose- and time-dependent decrease in cell viability, in both 5–8F
and 6–10B cells.
NaBu induces NPC cells apoptosis by
activating the mitochondrial pathway
Apoptotic induction is one of the common mechanisms
responsible for the anti-tumor roles of NaBu (22,23). In
the present study, obvious apoptosis was observed in 5–8F and 6–10B
cells, as demonstrated by flow cytometry analysis (Fig. 2A and B). In accord with the results of
the MTT assay, the apoptotic rate of NPC cells treated with NaBu
also exhibited a dose- and time-dependent pattern (Fig. 2A and B). Subsequently, to identify the
underlying mechanisms, the expression of proteins involved in the
mitochondrial apoptosis pathway, an important signaling axis in
apoptosis regulation, were detected. Downregulated expression of
anti-apoptotic proteins, including Bcl-2, Bcl-xL, Mcl1 and
survivin, was observed in 5–8F and 6–10B cells treated with 5 mM
NaBu (Fig. 2C). Bax, one of the
pro-apoptotic proteins, was downregulated in the NaBu-treated group
(Fig. 2C). Furthermore, c-Casp3 and
c-PARP, two of the apoptotic executors, were upregulated, while
Casp3 was correspondingly downregulated in the NaBu-treated group
(Fig. 2C). Taken together, these
results indicate that NaBu could induce the apoptosis of NPC cells
by activating the mitochondrial apoptosis pathway.
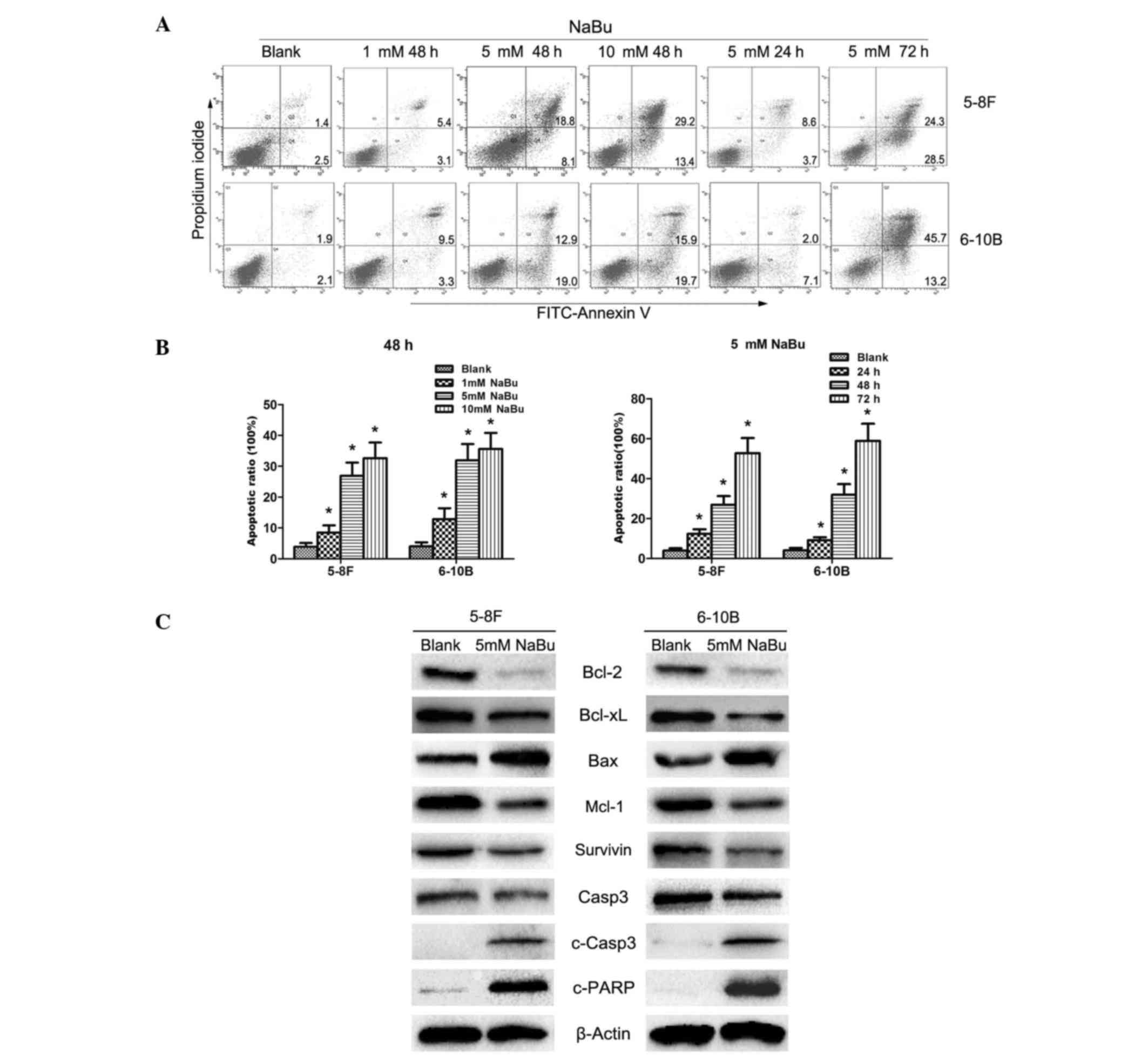 | Figure 2.NaBu induced nasopharyngeal carcinoma
cell apoptosis by activating the mitochondrial apoptotic axis. (A
and B) NaBu induced cell apoptosis in 5–8F and 6–10B cells in a
dose- and time-dependent manner *P<0.05 vs. blank. The numbers
in the quadrants correspond to percentages. (C) NaBu activated the
mitochondrial apoptosis axis in 5–8F and 6–10B cells. The
corresponding expression fluctuations of mitochondrial apoptotic
proteins, Bcl-2 (decreased), Bcl-xL (decreased), Mcl-1 (decreased),
survivin (decreased), Bax (increased), Casp3 (decreased), c-Casp3
(increased) and c-PARP (increased), confirmed the activation of the
mitochondrial apoptosis axis in 5–8F and 6–10B cells treated with
NaBu. NaBu, sodium butyrate; FITC, fluorescein isothiocyanate; Bcl,
B-cell lymphoma; xL, extra large; Bax, Bcl-2-associated X; Mcl1,
myeloid leukemia cell differentiation protein-1; c-, cleaved;
Casp3, caspase 3; PARP, poly ADP ribose polymerase. |
SOCE inhibition can attenuate the
apoptosis induced by NaBu
Ca2+, as a key important second message,
plays vital roles in cell signaling transduction processes such as
apoptosis regulation (13). NaBu
could activate the SOCE process in colon cancer cells to initiate
apoptosis (17). In the present
study, similar results were revealed in NPC cells. Influx of
extracellular Ca2+ was observed in 5–8F cells
immediately upon NaBu addition (Fig.
3), and similar outcomes were observed in 6–10B cells (data not
shown). The Ca2+ influx was inhibited when EGTA, a
Ca2+ chelator, or 2-APB, a SOCE inhibitor, were added
together with NaBu. Correspondingly, the apoptosis induced by NaBu
was attenuated by EGTA and 2-APB, as demonstrated by the lower
apoptotic rates of the EGTA/2-APB-treated group compared with those
of the NaBu alone-treated group (Fig. 4A
and B), and by the increased levels of Bcl-xL and decreased
levels of c-PARP in the EGTA/2-APB-treated group compared with
those in the NaBu alone-treated group in western blot analysis
(Fig. 4C). Therefore, the SOCE
process may be involved in the NaBu-induced apoptosis of NPC
cells.
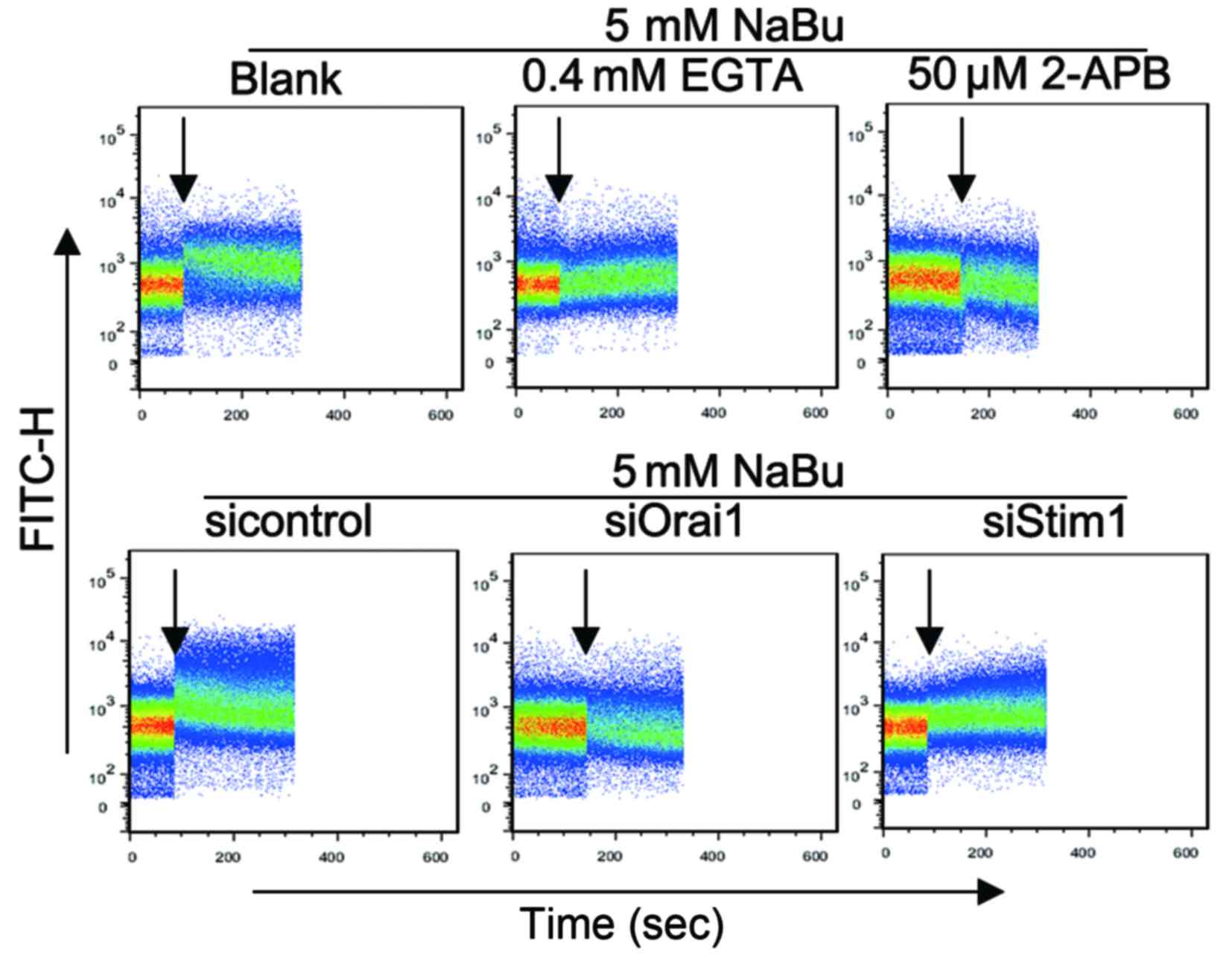 | Figure 3.NaBu-induced Ca2+ influx
in 5–8F and 6–10B cells by increased SOCE. NaBu addition could
promote Ca2+ influx immediately in 5–8F cells.
NaBu-induced Ca2+ influx could be attenuated by the SOCE
inhibitor 2-APB, the Ca2+ chelator EGTA, and
downregulation of Orai1 and Stim1. The black arrows indicate the
time of agents addition. FITC, fluorescein isothiocyanate; NaBu,
sodium butyrate; Orai1, calcium release-activated calcium channel
protein 1; Stim1, stromal interaction molecule 1; si, small
interfering; 2-APB, 2-aminoethoxydiphenyl borate; EGTA,
glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid; SOCE,
store-operated Ca2 entry. |
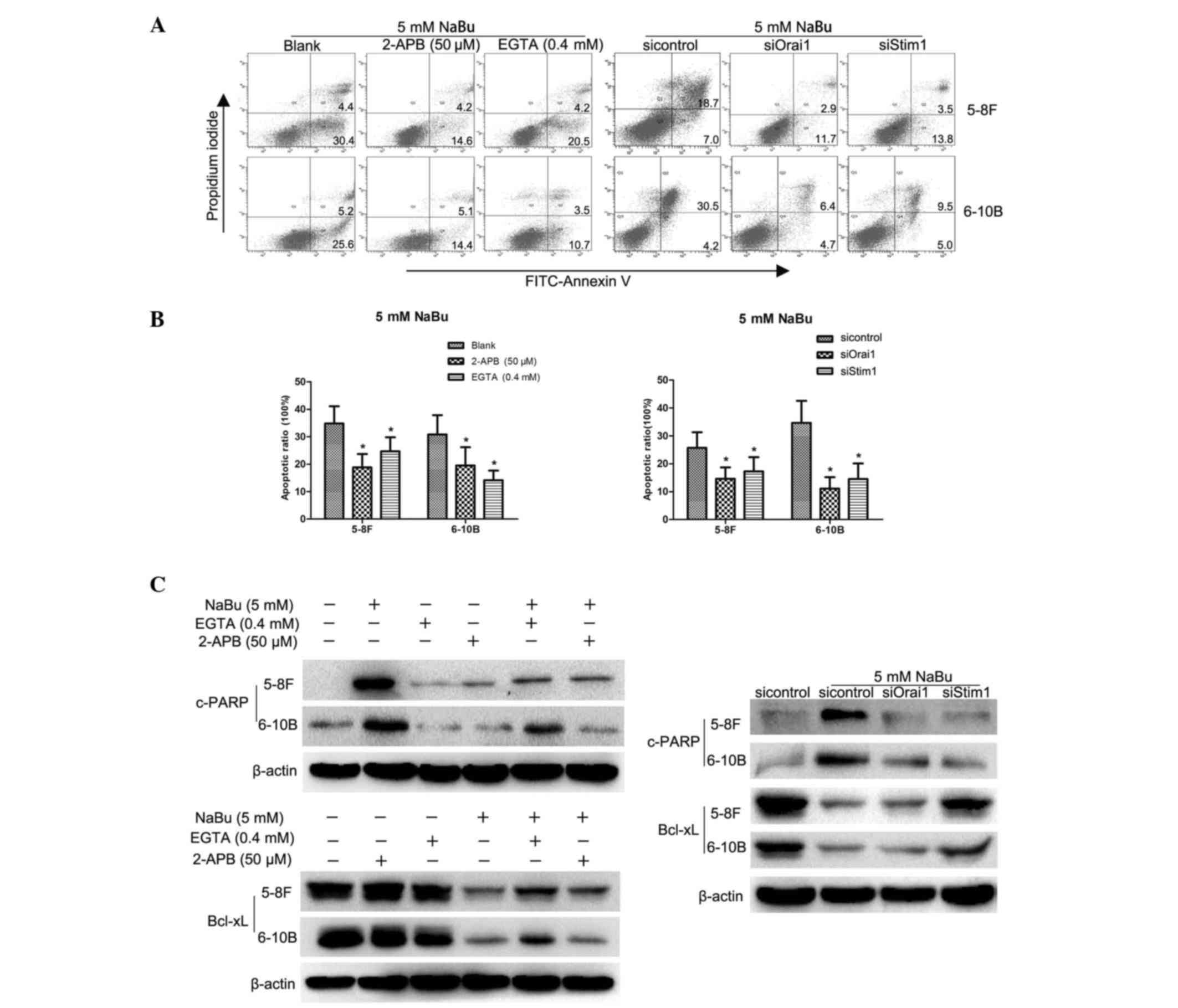 | Figure 4.Inhibition of store-operated
Ca2+ entry could attenuate the apoptosis induced by NaBu
in NPC cells. (A and B) Flow cytometry analyses revealed that the
NaBu-induced apoptotic rates were decreased by 2-APB, EGTA, and
downregulation of Orai1 and Stim1. *P<0.05, experimental groups
compared with blank groups. The numbers in the quadrants correspond
to percentages. (C) Western blot analysis demonstrated that
NaBu-induced apoptosis was attenuated, as demonstrated by the
lesser extent of fluctuations in Bcl-xL and c-PARP expression
levels in NPC cells with downregulated Orai1 and Stim1 expression
compared with those in the control group. NPC, nasopharyngeal
carcinoma; FITC, fluorescein isothiocyanate; NaBu, sodium butyrate;
Orai1, calcium release-activated calcium channel protein 1; Stim1,
stromal interaction molecule 1; si, small interfering; 2-APB,
2-aminoethoxydiphenyl borate; EGTA, glycol-bis(β-aminoethyl
ether)-N,N,N',N'-tetraacetic acid; c-PAPR, cleaved poly ADP ribose
polymerase; Bcl-xL, B-cell lymphoma-extra large. |
Disruption of the CRAC channel can
also attenuate the apoptosis induced by NaBu
The CRAC channel is one of the major pathways of
SOCE progression (14). The present
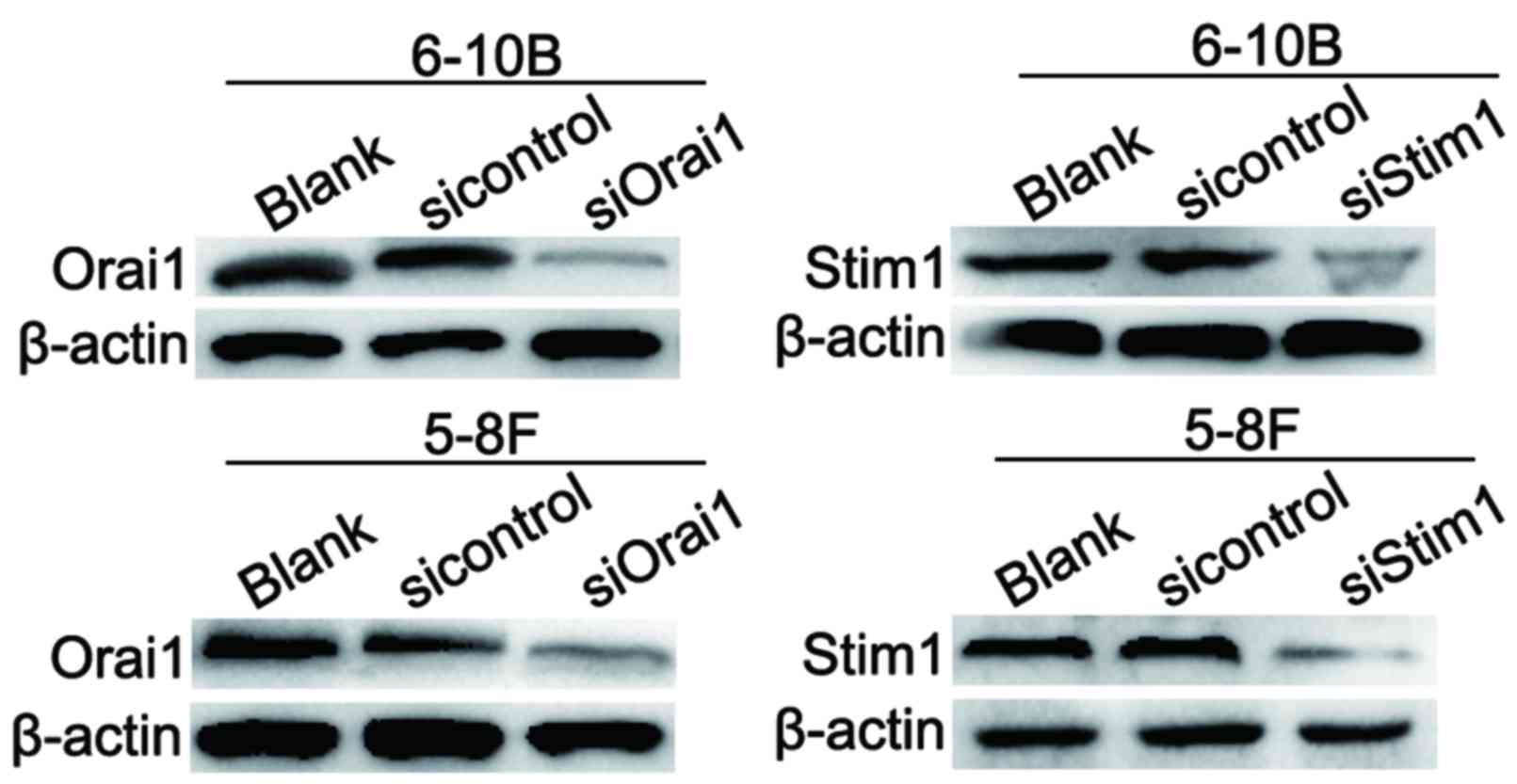
study successfully knocked down the expression of Orai1 and Stim1,
two key components of the CRAC channel, in 5–8F and 6–10B cells
(Fig. 5). Then, the Ca2+
influx induced by NaBu was analyzed, and it was observed that the
Ca2+ influx was attenuated in 5–8F cells with
downregulated Orai1 and Stim1 expression (Fig. 3). Similar results were obtained in
6–10B cells with downregulated Orai1 and Stim1 expression (data not
shown). The apoptotic rates were decreased in NPC cells with
downregulated Orai1 and Stim1 expression compared with those in the
control group (Fig. 4A and B).
Furthermore, the extent of fluctuations in Bcl-xL and c-PARP
expression levels were decreased in NPC cells with downregulated
Orai1 and Stim1 expression compared with those in the control group
(Fig. 4C). Thus, based on the these
results, the SOCE process appears to be necessary for the apoptosis
induced by NaBu in NPC cells.
Discussion
It is well known that epigenetic aberrations
contribute substantially to the onset and progression of human
diseases, particularly cancers. HDACs, which are key epigenetic
regulators, have been demonstrated to be involved in cancer
progression (6,8). Overexpression of HDACs displayed a
negative correlation with disease-free survival and OS, and could
serve as a biomarker for prognosis prediction in multiple cancer
types, including prostate (24),
colorectal (25) and breast cancer
(26). Thus, HDACis, which can
inhibit the enzymatic activities of HDACs, are promising agents for
cancer treatment. Currently, three HDACis, vorinostat, romidepsin
and belinostat, have been approved for the treatment of cutaneous
T-cell lymphoma and peripheral T-cell lymphoma (8).
NaBu, as a potent HDACi, has been confirmed to be
capable of exerting anti-tumor roles in various cancers. For
example, NaBu treatment could induce growth inhibition and
morphological transformation in prostate cancer (21,23),
breast cancer (27) and colorectal
cancer (9). NaBu has been
demonstrated to inhibit interferon-γ-induced indoleamine
2,3-dioxygenase expression in NPC cells (28). However, its cytotoxicity towards NPC
cells was rarely studied. In the present study, NaBu exhibited
significant cytotoxicity in NPC cells in a dose- and time-dependent
manner, and inhibited their proliferation ability. Furthermore,
NaBu treatment also induced morphological changes in NPC cells.
Consequently, NaBu was concluded to be cytotoxic to NPC cells, in
accordance with its known functions in other cancers.
Apoptosis induction is one of the common mechanisms
of anti-tumor agents (29). Apparent
cell apoptosis both through the death receptor (extrinsic)- and the
mitochondrion (intrinsic)-mediated pathways, has been observed in
multiple cancer cells treated with NaBu (9,22,30). For example, NaBu can activate
mitochondrial apoptosis, as demonstrated by the expression changes
in the Bcl-2 family of proteins, including upregulation of the
pro-apoptotic Bcl-2 homologous antagonist/killer and Bax, and
downregulation of the anti-apoptotic Bcl-2 and Bcl-xL (9). In the present study, flow cytometry
analysis revealed that NaBu can also induce significant apoptosis
in NPC cells. Furthermore, the decreased expression of
anti-apoptotic proteins, including Bcl-2, Bcl-xL, Mcl-1 and
survivin, in combination with the increased expression of
pro-apoptotic proteins such as Bax and apoptotic executors such as
c-Casp3 and c-PARP, confirmed the activation of the mitochondrial
apoptosis axis in NPC cells treated with NaBu. Therefore, the
present results revealed that the activation of mitochondrial
apoptosis involves the anti-tumor roles of NaBu in NPC.
The important roles of Ca2+ and its ion
channels in apoptotic regulation have been illustrated in numerous
studies (31–33). A sustained increase in cytosolic
Ca2+ activity triggers apoptosis (34). The roles of SOCE, a major
calcium-entry pathway for non-excitable cells, and those of the
CRAC channel, a key channel in mediating SOCE, in apoptotic
regulation appear to be paradoxical (7,16,17,34,35).
Indeed, SOCE can serve as a pro-apoptotic or an anti-apoptotic
factor in cancerous cells under different conditions (7,16,17,34,35). For
example, enhanced SOCE resulted from upregulated Orai1 and Stim1
expression, which was observed in drug-resistant cancer cells, and
impairing SOCE by using specific inhibitors or by knocking down
Orai1 and Stim1 can sensitize the drug-resistant cells to
chemotherapy (34,35). In addition, enhanced SOCE commonly
occurs during the cell apoptosis induced by anti-tumor or
chemotherapeutic agents (17). The
use of SOCE inhibitors or knocking down the expression of Orai1 or
Stim1 can counteract the apoptosis induced by anti-tumor agents
(7,16,17). In
the present study, SOCE appeared to serve as a pro-apoptotic factor
in NPC cells treated with NaBu, and inhibition of SOCE could
attenuate the NaB-induced apoptosis in NPC cells, in accordance
with previous results in colon cancer (17). Differences of SOCE in apoptosis may
contribute to the dual roles of Ca2+ in signaling
transduction: On one hand, a physiological Ca2+
concentration is necessary to activate signaling pathways that
promote cell growth, survival and metastasis, which are the
hallmarks of cancers; on the other hand, overloaded Ca2+
concentrations would also induce apoptosis through the activation
of apoptotic pathways (12,13,31,33).
Therefore, the anti-apoptotic role of enhanced SOCE in untreated
cancer cells is to maintain their own characteristics, which must
be regulated and controlled by upstream proteins (13,34).
However, the enhanced SOCE observed in the pro-apoptosis of cancer
cells treated with anti-tumor agents may be due to the impaired
upstream control signaling disrupted by anti-tumor agents, which
leads to a sustained increase in intracellular Ca2+
levels and triggers apoptosis.
In conclusion, the present study demonstrated that
NaBu was able to trigger hyperactivated SOCE and subsequently
induce the mitochondrial apoptosis pathway, which may underlie the
mechanisms of its cytotoxicity on NPC cells. The results of the
present study suggested that HDACis like NaBu may serve as
promising chemotherapeutic or adjuvant chemotherapeutic agents in
NPC therapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (Beijing, China; grant no.
81272972), the National Basic Research Program of China (Beijing,
China; grant no. 2010CB833605), the Incubation Program for National
Natural Science Funds for Distinguished Young Scholar of Central
South University (Changsha, China; grant no. 2010QYZD006), the
Hunan Provincial Science and Technology Department (Changsha,
China; grant no. 2013FJ4010), the National Science Foundation of
Hunan Province (grant no. 2016JJ2172) and the Open-End Fund for the
Valuable and Precision Instruments of Central South University
(Changsha, China; grant nos. CSUZC201634 and CSUZC201638).
References
|
1
|
Cao SM, Simons MJ and Qian CN: The
prevalence and prevention of nasopharyngeal carcinoma in China.
Chin J Cancer. 30:114–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feng X, Ren C, Zhou W, Liu W, Zeng L, Li
G, Wang L, Li M, Zhu B, Yao K and Jiang X: Promoter
hypermethylation along with LOH, but not mutation, contributes to
inactivation of DLC-1 in nasopharyngeal carcinoma. Mol Carcinog.
53:858–870. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stoker SD, van Diessen JN, de Boer JP,
Karakullukcu B, Leemans CR and Tan IB: Current treatment options
for local residual nasopharyngeal carcinoma. Curr Treat Options
Oncol. 14:475–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang L, Chen QY, Liu H, Tang LQ and Mai
HQ: Emerging treatment options for nasopharyngeal carcinoma. Drug
Des Devel Ther. 7:37–52. 2013.PubMed/NCBI
|
|
5
|
Colaco RJ, Betts G, Donne A, Swindell R,
Yap BK, Sykes AJ, Slevin NJ, Homer JJ and Lee LW: Nasopharyngeal
carcinoma: A retrospective review of demographics, treatment and
patient outcome in a single centre. Clin Oncol (R Coll Radiol).
25:171–177. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Falkenberg KJ and Johnstone RW: Histone
deacetylases and their inhibitors in cancer, neurological diseases
and immune disorders. Nat Rev Drug Discov. 13:673–691. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun DP, Li XX, Liu XL, Zhao D, Qiu FQ, Li
Y and Ma P: Gypenosides induce apoptosis by ca2+ overload mediated
by endoplasmic-reticulum and store-operated ca2+ channels in human
hepatoma cells. Cancer Biother Radiopharm. 28:320–326. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Goncalves P and Martel F: Butyrate and
colorectal cancer: The role of butyrate transport. Curr Drug Metab.
14:994–1008. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tailor D, Hahm ER, Kale RK, Singh SV and
Singh RP: Sodium butyrate induces DRP1-mediated mitochondrial
fusion and apoptosis in human colorectal cancer cells.
Mitochondrion. 16:55–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei ZL, Zhao QL, Yu DY, Hassan MA, Nomura
T and Kondo T: Enhancement of sodium butyrate-induced cell death
and apoptosis by X-irradiation in the human colorectal cancer cell
line HCT 116. Oncol Rep. 20:397–403. 2008.PubMed/NCBI
|
|
12
|
Prevarskaya N, OuadidAhidouch H, Skryma R
and Shuba Y: Remodelling of Ca2+ transport in cancer: How it
contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol
Sci. 369:201300972014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen YF, Chen YT, Chiu WT and Shen MR:
Remodeling of calcium signaling in tumor progression. J Biomed Sci.
20:232013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bergmeier W, Weidinger C, Zee I and Feske
S: Emerging roles of store-operated Ca2+ entry through
STIM and ORAI proteins in immunity, hemostasis and cancer. Channels
(Austin). 7:379–391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chiu WT, Tang MJ, Jao HC and Shen MR: Soft
substrate up-regulates the interaction of STIM1 with store-operated
Ca2+ channels that lead to normal epithelial cell apoptosis. Mol
Biol Cell. 19:2220–2230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Flourakis M, Lehen'kyi V, Beck B, Raphaël
M, Vandenberghe M, Abeele FV, Roudbaraki M, Lepage G, Mauroy B,
Romanin C, et al: Orai1 contributes to the establishment of an
apoptosis-resistant phenotype in prostate cancer cells. Cell Death
Dis. 1:e752010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun S, Li W, Zhang H, Zha L, Xue Y, Wu X
and Zou F: Requirement for store-operated calcium entry in sodium
butyrate-induced apoptosis in human colon cancer cells. Biosci Rep.
32:83–90. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang S, Zhang JJ and Huang XY: Orai1 and
STIM1 are critical for breast tumor cell migration and metastasis.
Cancer Cell. 15:124–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Feng X, Li C, Liu W, Chen H, Zhou W, Wang
L, Zhu B, Yao K, Jiang X and Ren C: DLC-1, a candidate tumor
suppressor gene, inhibits the proliferation, migration and
tumorigenicity of human nasopharyngeal carcinoma cells. Int J
Oncol. 42:1973–1984. 2013.PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Paskova L, Trtkova K Smesny, Fialova B,
Benedikova A, Langova K and Kolar Z: Different effect of sodium
butyrate on cancer and normal prostate cells. Toxicol in vitro.
27:1489–1495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maruyama T, Yamamoto S, Qiu J, Ueda Y,
Suzuki T, Nojima M and Shima H: Apoptosis of bladder cancer by
sodium butyrate and cisplatin. J Infect Chemother. 18:288–295.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mu D, Gao Z, Guo H, Zhou G and Sun B:
Sodium butyrate induces growth inhibition and apoptosis in human
prostate cancer DU145 cells by up-regulation of the expression of
annexin A1. PloS One. 8:e749222013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weichert W, Röske A, Gekeler V, Beckers T,
Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M
and Kristiansen G: Histone deacetylases 1, 2 and 3 are highly
expressed in prostate cancer and HDAC2 expression is associated
with shorter PSA relapse time after radical prostatectomy. Br J
Cancer. 98:604–610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weichert W, Röske A, Niesporek S, Noske A,
Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T and Denkert
C: Class I histone deacetylase expression has independent
prognostic impact in human colorectal cancer: Specific role of
class I histone deacetylases in vitro and in vivo. Clin Cancer Res.
14:1669–1677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Krusche CA, Wülfing P, Kersting C, Vloet
A, Böcker W, Kiesel L, Beier HM and Alfer J: Histone deacetylase-1
and −3 protein expression in human breast cancer: A tissue
microarray analysis. Breast Cancer Res Treat. 90:15–23. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun B, Liu R, Xiao ZD and Zhu X: c-MET
protects breast cancer cells from apoptosis induced by sodium
butyrate. PloS One. 7:e301432012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He YW, Wang HS, Zeng J, Fang X, Chen HY,
Du J and Yang XY: Sodium butyrate inhibits interferon-gamma induced
indoleamine 2,3-dioxygenase expression via STAT1 in nasopharyngeal
carcinoma cells. Life sci. 93:509–515. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gospodinov A, Popova S, Vassileva I and
Anachkova B: The inhibitor of histone deacetylases sodium butyrate
enhances the cytotoxicity of mitomycin C. Mol Cancer Ther.
11:2116–2126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pinton P, Giorgi C, Siviero R, Zecchini E
and Rizzuto R: Calcium and apoptosis: ER-mitochondria Ca2+ transfer
in the control of apoptosis. Oncogene. 27:6407–6418. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rizzuto R, Pinton P, Ferrari D, Chami M,
Szabadkai G, Magalhães PJ, Di Virgilio F and Pozzan T: Calcium and
apoptosis: Facts and hypotheses. Oncogene. 22:8619–8627. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Prevarskaya N, Skryma R and Shuba Y:
Targeting Ca2+ transport in cancer: Close reality or long
perspective? Expert Opin Ther Targets. 17:225–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schmidt S, Liu G, Liu G, Yang W, Honisch
S, Pantelakos S, Stournaras C, Hönig A and Lang F: Enhanced Orai1
and STIM1 expression as well as store operated Ca2+ entry in
therapy resistant ovary carcinoma cells. Oncotarget. 5:4799–4810.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kondratska K, Kondratskyi A, Yassine M,
Lemonnier L, Lepage G, Morabito A, Skryma R and Prevarskaya N:
Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance
of pancreatic adenocarcinoma. Biochim Biophys Acta. 1843:2263–2269.
2014. View Article : Google Scholar : PubMed/NCBI
|