Introduction
Gastric cancer (GC) is one of the most common
malignancies worldwide and a leading cause of cancer-related
mortality (1,2). Little is known about GC-associated genes
or its diverse clinical properties, including metastatic status,
invasiveness, histological type and responsiveness to chemotherapy
(3). The carcinogenic mechanism of GC
is still not fully understood, although previous studies have
demonstrated a number of genetic alterations in this disease
(3).
Studies have revealed that trichostatin A (TSA) has
an inhibitory role in GC (4–7). In eukaryotic transcription, chromatin
modifications by histone acetyltransferase or histone
deacetyltransferase (HDAC) represent a fundamental mechanism of
transcriptional regulation (8).
Evidence from further studies suggests that histone acetylation
alters nucleosomal structures and facilitates the accessibility of
transcription-associated factors to chromatin DNA through the
disruption of interactions between histones and DNA (9–11). As an
HDAC inhibitor, TSA has been known to cause a variety of phenotypic
changes, including cell-cycle arrest in the G1/G2 phase, apoptosis
and differentiation in cultured transformed cells (12–14).
Although significant efforts have been made, the candidate genes
and inhibition mechanism of TSA in GC remain unclear.
In the present study, microarray data were obtained
through chip detection, and the differentially expressed genes
(DEGs) between TSA-treated GC cells and untreated GC cells were
identified. In addition, gene ontology (GO) analysis of the DEGs
was performed. Then sub-pathway enrichment analysis was performed,
and a microRNA (miRNA) regulatory network was constructed. Through
the identification of GC-associated genes and biological changes,
possible molecular mechanisms and potential therapeutic targets for
GC were explored.
Materials and methods
Affymetrix microarray data
The GC cell line, BGC-823, was grown in RPMI-1640
supplemented with 10% fetal bovine serum and antibiotics in 5%
CO2. The cells were grown to ~70–80% confluence and
treated with 330 nM TSA (Sigma-Aldrich, St. Louis, MO, USA) for 24
h. Then total RNA was isolated from TSA-treated cells using TRIzol
(Invitrogen Life Technologies, Gaithersburg, MD, USA). The
construction of a fluorescence probe and hybridization were
performed using a 3DNA array detection kit (Genisphere, Hatfield,
PA, USA). cDNA microarrays were performed on a cDNA chip containing
21,522 cDNA clones selected from the Human Genome Oligo set version
2.1 (Operon, Ebersberg, Germany). Each slide contained 12 control
genes to normalize the signal intensities of the different
fluorescent dyes. Each hybridization array was scanned on a LuxScan
10KA (CapitalBio, Beijing, China). The intensity of each
hybridization signal was calculated by the GenePix Pro 4.0 program
(Axon, Schaumburg, IL, USA). Three repeats were included in this
study, and each repeat contained TSA treated GC cells and untreated
cells.
Data processing and DEG analysis
The signal intensities of TSA treated GC cells and
untreated GC cells in each repeat were obtained through chip
detection and the gene expression ratio (TSA-treated GC cells /
untreated GC cells) in each repeat was calculated. Then, the DEGs
between the TSA-treated group and the control group were analyzed,
and the score was calculated by the significant analysis of
microarray (SAM) algorithm (15).
Each gene was assigned a difference score (defined as “d”) based on
the significance of its gene expression changes between TSA treated
GC cells and untreated GC cells. The multiple testing correction
was performed using a false discovery rate (FDR) (16). The fold change of the expression of
individual genes was also observed for the differential expression
test. DEGs with an FDR<0.05 were considered to be significant.
DEGs with a score of (d) ≥1.25 and a ratio (TSA-treated GC cells /
untreated GC cells) >1.5 in at least two groups were defined as
upregulated DEGs, and DEGs with a score of (d) ≤-1.25 and a ratio
(TSA-treated GC cells / untreated GC cells) <0.66 in at least
two groups were defined as downregulated DEGs.
GO analysis
GO analysis has become a commonly used approach for
functional studies of large-scale genomic or transcriptomic data
(17). The database for annotation,
visualization and integrated discovery (DAVID) (18) consists of an integrated biological
knowledge base and analytic tools aimed at systematically
extracting biological meaning from large gene or protein lists. The
GO function of DEGs was analyzed using DAVID. P<0.05 was
considered to indicate a statistically significant difference.
Sub-pathway enrichment analysis
The Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway database (19) contains
information on how molecules or genes are networked, and is
complementary to most of the existing molecular biological
databases that contain information on individual molecules or
genes. The KEGG pathways of the DEGs were analyzed using DAVID.
P<0.05 was considered to indicate a statistically significant
difference. The closer the components within the metabolic
pathways, the greater the similarity in their biological functions.
Therefore, the identification of the sub-pathway of disease is
crucial. K-clique was used to divide the metabolic pathway into
sub-pathways through the iSubpathwayMiner package in R (https://www.r-project.org/). The sub-pathway with
P<0.05 was considered to be significant.
miRNA regulatory network analysis
miRNAs are a type of endogenous non-coding RNA with
regulatory functions, and their size is ~20–25 nucleotides. Mature
miRNAs are assembled into RNA-induced silencing complexes, then
target genes are identified and regulated by them. Thus, the
identification of the miRNA regulatory network is essential. We
predicted the interactions between target genes and miRNAs based on
the TarBase (http://microrna.gr/tarbase) (20), TargetScan (www.targetscan.org) (21) and miRecord (http://mire-cords.biolead.org) (22) databases. The miRNA regulatory network
was constructed using Cytoscape (http://cytoscape.org/) (23).
Literature mining analysis of key
genes
The online tool GenCLip2.0 was used to mine DEGs in
literature (24). A list of DEG names
was input into GenCLip2.0, then the Gene Cluster with Literature
Profiles module was used to cluster based on the documented
frequency of the input genes. GenCLip2.0 also provided GO and
pathway enrichment analysis of genes. The GC research-related DEGs
were mined from the literature and these DEGs were submitted to GO
and pathway enrichment analyses.
Results
Screening and GO analysis of DEGs
Three groups of gene expression ratio (TSA-treated
GC cells / untreated GC cells) about TSA-treated group and control
group were obtained through chip detection. A total of 76 DEGs (43
downregulated genes and 33 upregulated genes) were obtained. The
results of GO analysis revealed that the upregulated DEGs were
mainly enriched in biological processes including skeletal muscle
contraction, response to organic substance and regulation of cell
adhesion, and the downregulated DEGs were mainly enriched in
processes including the DNA catabolic process, DNA metabolic
process and response to the tumor necrosis factor (Table I).
 | Table I.Gene ontology analysis results of
differentially expressed genes. |
Table I.
Gene ontology analysis results of
differentially expressed genes.
| Gene type | Category | Term | Count | P-value |
|---|
| Upregulated | GOTERM_BP_FAT |
GO:0003010~voluntary skeletal muscle
contraction | 2 | 0.004135 |
|
| GOTERM_BP_FAT | GO:0014721~twitch
skeletal muscle contraction | 2 | 0.004135 |
|
| GOTERM_BP_FAT |
GO:0031444~slow-twitch skeletal muscle
fiber contraction | 2 | 0.004135 |
|
| GOTERM_BP_FAT | GO:0010033~response
to organic substance | 6 | 0.015009 |
|
| GOTERM_BP_FAT | GO:0003009~skeletal
muscle contraction | 2 | 0.024566 |
|
| GOTERM_BP_FAT |
GO:0030155~regulation of cell
adhesion | 3 | 0.032410 |
|
| GOTERM_BP_FAT |
GO:0050881~musculoskeletal movement | 2 | 0.038627 |
|
| GOTERM_BP_FAT |
GO:0050879~multicellular organismal
movement | 2 | 0.038627 |
|
| GOTERM_BP_FAT | GO:0009725~response
to hormone stimulus | 4 | 0.039305 |
|
| GOTERM_BP_FAT | GO:0050873~brown
fat cell differentiation | 2 | 0.048550 |
|
| GOTERM_CC_FAT |
GO:0005576~extracellular region | 10 | 0.010773 |
|
| GOTERM_CC_FAT |
GO:0005615~extracellular space | 5 | 0.042191 |
|
| GOTERM_MF_FAT |
GO:0003735~structural constituent of
ribosome | 3 | 0.041104 |
| Downregulated | GOTERM_BP_FAT | GO:0006308~DNA
catabolic process | 3 | 0.004610 |
|
| GOTERM_BP_FAT | GO:0006259~DNA
metabolic process | 5 | 0.009728 |
|
| GOTERM_BP_FAT |
GO:0006301~post-replication repair | 2 | 0.013524 |
|
| GOTERM_BP_FAT | GO:0034612~response
to tumor necrosis factor | 2 | 0.021888 |
|
| GOTERM_BP_FAT | GO:0051129~negative
regulation of cellular component organization | 3 | 0.023964 |
|
| GOTERM_BP_FAT |
GO:0065003~macromolecular complex
assembly | 5 | 0.024316 |
|
| GOTERM_BP_FAT | GO:0006974~response
to DNA damage stimulus | 4 | 0.024466 |
|
| GOTERM_BP_FAT | GO:0070647~protein
modification by small protein conjugation or removal | 3 | 0.029893 |
|
| GOTERM_BP_FAT |
GO:0043933~macromolecular complex subunit
organization | 5 | 0.030046 |
|
| GOTERM_BP_FAT | GO:0006309~DNA
fragmentation involved in apoptosis | 2 | 0.031835 |
|
| GOTERM_BP_FAT | GO:0044265~cellular
macromolecule catabolic process | 5 | 0.032124 |
|
| GOTERM_BP_FAT | GO:0006921~cell
structure disassembly during apoptosis | 2 | 0.038412 |
|
| GOTERM_BP_FAT |
GO:0009057~macromolecule catabolic
process | 5 | 0.040630 |
|
| GOTERM_BP_FAT | GO:0000737~DNA
catabolic process, endonucleolytic | 2 | 0.043317 |
|
| GOTERM_BP_FAT |
GO:0030262~apoptotic nuclear changes | 2 | 0.043317 |
|
| GOTERM_BP_FAT | GO:0009314~response
to radiation | 3 | 0.044895 |
|
| GOTERM_CC_FAT |
GO:0043232~intracellular
non-membrane-bounded organelle | 9 | 0.025314 |
|
| GOTERM_CC_FAT |
GO:0043228~non-membrane-bounded
organelle | 9 | 0.025314 |
|
| GOTERM_CC_FAT |
GO:0070013~intracellular organelle
lumen | 7 | 0.038954 |
|
| GOTERM_CC_FAT |
GO:0043233~organelle lumen | 7 | 0.042966 |
|
| GOTERM_CC_FAT |
GO:0031974~membrane-enclosed lumen | 7 | 0.046708 |
Sub-pathway enrichment analysis
The sub-pathway of disease was screened. The
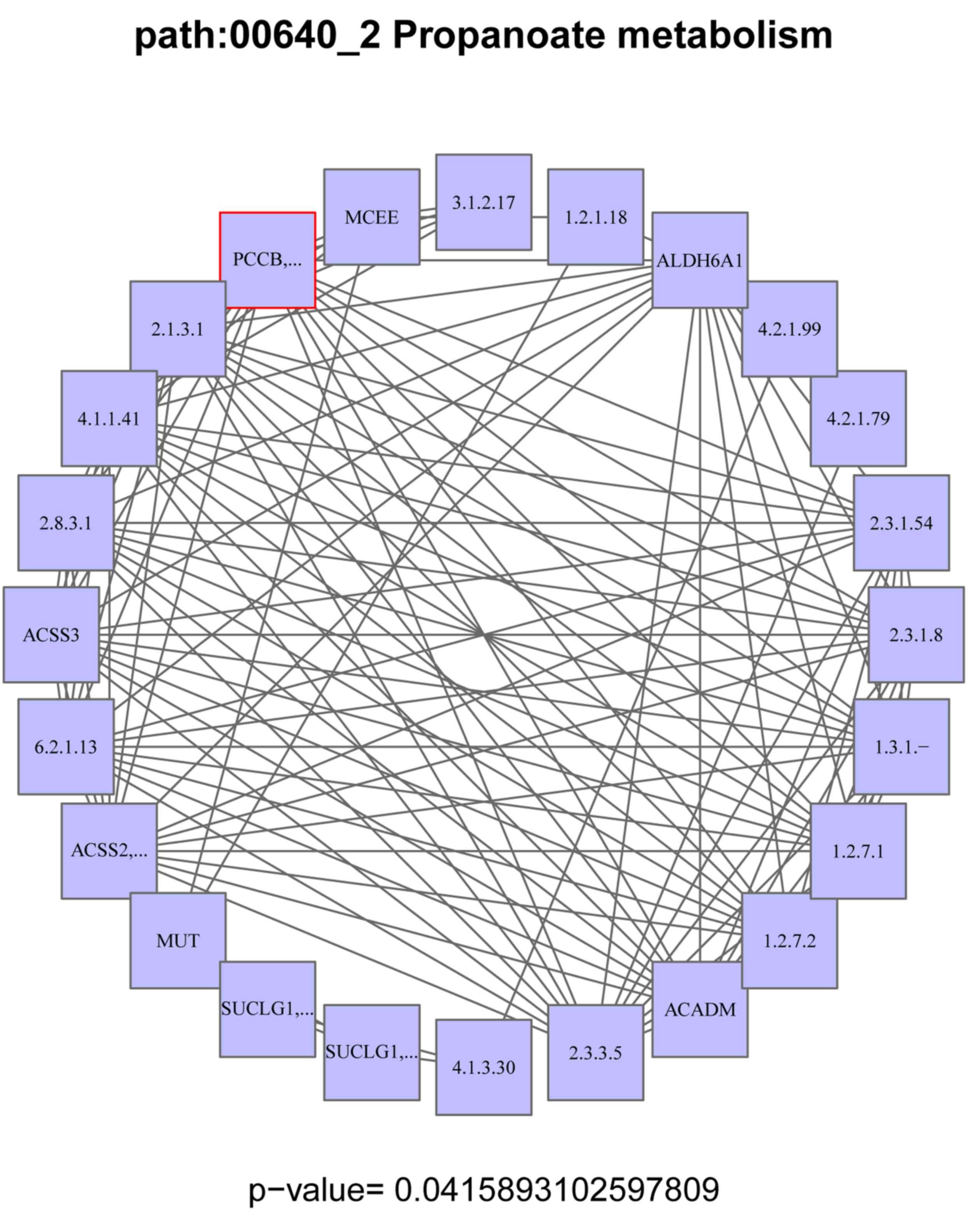
propanoate metabolism pathway was the sub-pathway selected in our
study. Propionyl-coenzyme A carboxylase, beta polypeptide
(PCCB) was the DEG enriched in this sub-pathway (Fig. 1).
miRNA regulatory network analysis
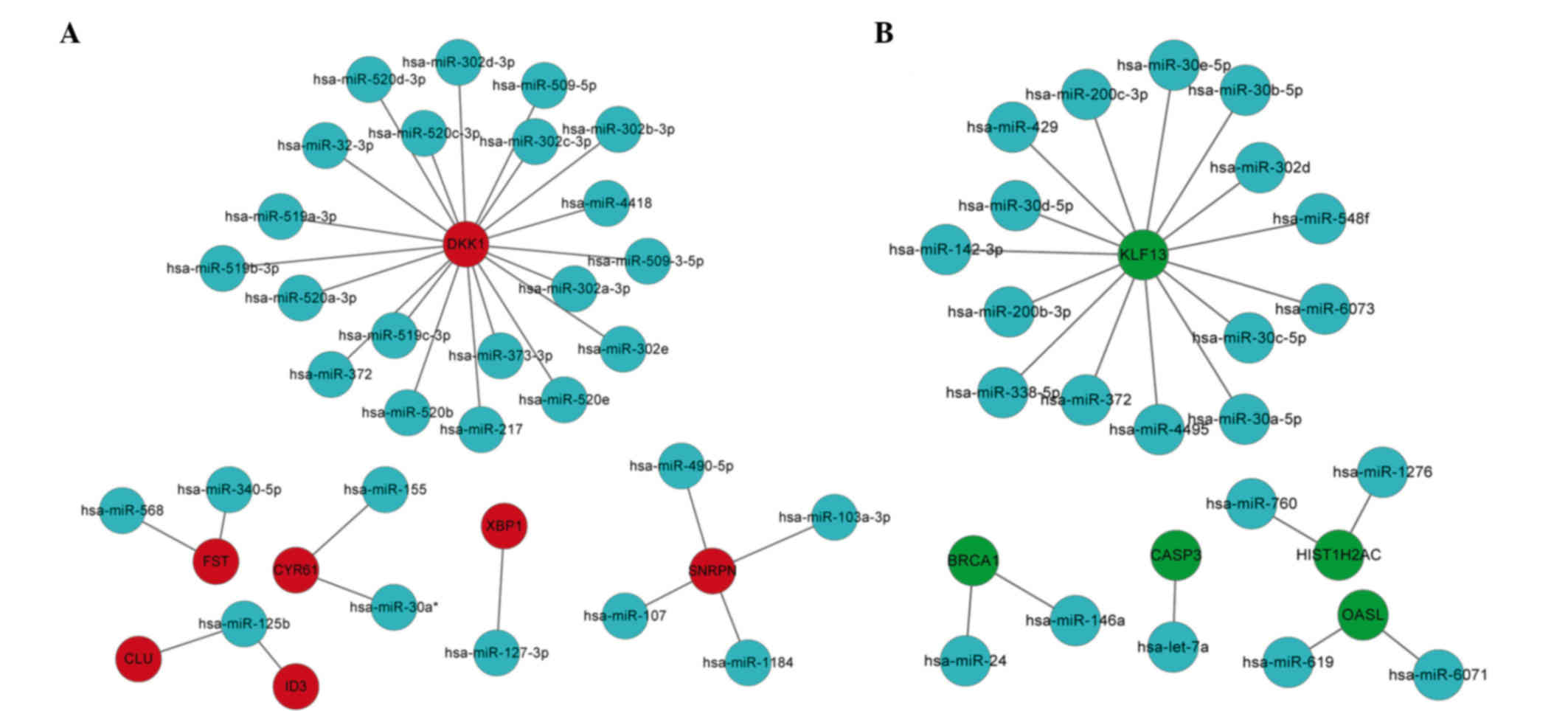
According to the miRNA regulatory network, the
upregulated genes dickkopf wnt signaling pathway inhibitor 1
(DKK1), follistatin (FST), clusterin (CLU),
inhibitor of DNA binding 3, dominant negative helix-loop-helix
protein (ID3), cysteine-rich, angiogenic inducer 61
(CYR61), x-box binding protein 1 (XBP1) and small
nuclear ribonucleoprotein polypeptide N (SNRPN) were the hub
nodes, and among them DKK1 was the top hub node (Fig. 2A). From the miRNA regulatory network,
the downregulated genes kruppel-like factor 13 (KLF13),
breast cancer 1, early onset (BRCA1), caspase 3,
apoptosis-related cysteine peptidase (CASP3),
2′-5′-oligoadenylate synthetase-like (OASL) and histone
cluster 1, H2ac (HIST1H2AC) were the hub nodes, and among
them KLF13 was the top hub node (Fig. 2B).
Literature mining analysis of key
genes
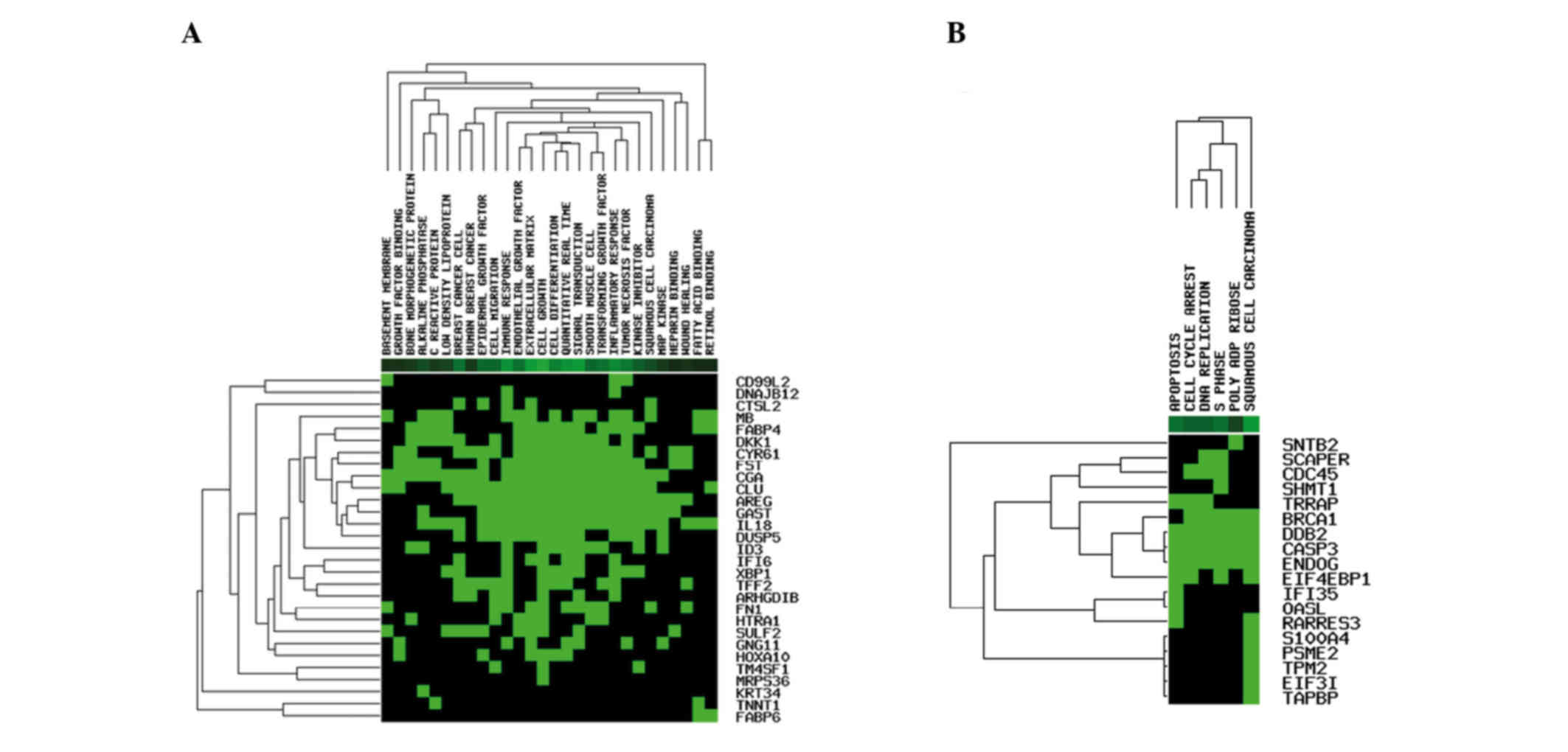
A heatmap of DEGs was constructed (Fig. 3). From the heatmap, the upregulated
genes were mainly enriched in functions including epidermal growth
factor, inflammation and tumor necrosis factor; the downregulated
genes were mainly enriched in biological processes including
squamous cell carcinoma entries, apoptosis and the DNA replication
process. The results of GO analysis revealed that the upregulated
genes were mainly enriched in functions including extracellular
space and response to organic substance (Table II); the downregulated genes were
mainly enriched in the DNA metabolic process (Table II).
| Table II.Gene ontology analysis results of the
key genes. |
Table II.
Gene ontology analysis results of the
key genes.
| Gene type | Pathway | Hit enrichment
score | P-value | Q-value | GO ID |
|---|
| Upregulated | Cluster1 | 4.97 |
|
|
|
|
| Extracellular
space | 8.00 | 4.72E-06 | 0.002702511 | GO:0005615 |
|
| Extracellular
region | 13.00 | 2.42E-05 | 0.004620421 | GO:0005576 |
|
| Single1 | 4.90 |
|
|
|
|
| Response to organic
substance | 13.00 | 1.27E-05 | 0.003630953 | GO:0010033 |
|
| Single2 | 4.13 |
|
|
|
|
| BMP signaling
pathway | 4.00 | 7.42E-05 | 0.010635069 | GO:0030509 |
| Downregulated | Single1 | 6.14 |
|
|
|
|
| DNA metabolic
process | 8.00 | 7.25E-07 | 0.000332722 | GO:0006259 |
Discussion
GC is one of the most common malignant diseases and
a leading cause of cancer-related mortality worldwide (1,2). The
mechanism of TSA inhibition in GC is unclear. In the present study,
we used bioinformatics approaches to explore the candidate genes
induced by TSA in GC, and identified 76 DEGs between TSA-treated GC
cells and untreated GC cells. Among these DEGs, 43 genes were
upregulated and 33 were downregulated. In addition, sub-pathway
enrichment analysis was performed, and the propanoate metabolism
sub-pathway was identified. Then the miRNA regulatory network was
constructed; from this, DKK1 was identified as the top hub
node among the upregulated DEGs, while KLF13 was the top hub
node among the downregulated DEGs.
The propanoate metabolism pathway was the
sub-pathway enriched in our study. To date, there have been no
studies on the correlation between the propanoate metabolism
pathway and GC. However, certain studies have reported that this
sub-pathway is associated with other cancer types. Zhu et al
observed that bladder cancer-related long noncoding RNAs (lncRNAs)
demonstrated a significant association with the propanoate
metabolism pathway (25).
Furthermore, certain studies have reported that dysregulation of
lncRNAs is a primary feature of several human cancers, including
prostate cancer, breast cancer, GC, bladder cancer and
hepatocellular carcinoma (26–32). The
kidney cancer-related proteins were demonstrated to be involved
with a high degree of confidence in propanoate metabolism, pyruvate
metabolism, the urea cycle and arginine pathways (33). Gmeiner et al observed that the
downregulated DEGs in colon cancer were involved with propanoate
metabolism and the isoleucine degradation pathway (34). Yang et al revealed that valine
relating to lung cancer was involved in a number of metabolic
processes including the propanoate metabolism process (35). Gonzalez-Angulo et al
demonstrated that the metabolic pathways (glutamate metabolism,
chondroitin sulfate biosynthesis and propanoate metabolism) were
the most consistently upregulated in basal-like residual cancers
(36). Considering the studies above,
the propanoate metabolism pathway may play a significant role in
TSA inhibition in GC, and may be a therapeutic target for GC.
DKK1 and KLF13 were observed to be hub
nodes in our miRNA regulatory networks. We revealed that
DKK1 was an upregulated DEG. Hirata et al suggested
that DKK1 induced cell apoptosis and inhibited cell growth
in renal cell carcinoma (RCC) (37).
The levels of DKK1 were decreased in RCC cell lines, but
they increased following treatment with TSA. These authors also
revealed that TSA induced histone acetylation at the DKK1
promoter, which resulted in the reactivation of DKK1
expression in human RCC. This result demonstrated that the
silencing of DKK1 is caused by histone modification
(37). Lee et al demonstrated
that DKK1 was repressed by histone deacetylation in cervical
cancer cells (38). The DKK family
was demonstrated to suppress cell growth and induce cell apoptosis
in gastrointestinal and colon cancer cells (39,40).
Studies have revealed that DKK1 is preferentially expressed
in lung cancer, and it was identified as a novel prognostic
biomarker as well as a therapeutic target for lung cancers
(41,42). Certain studies demonstrated that DKK1
significantly reduced tumor growth (39,43).
DKK1 levels may aid in diagnosing GC, and the gene may be a
novel prognostic marker for GC (35).
Our results revealed that DKK1 was an upregulated DEG in GC
cells treated with TSA. Furthermore, the literature mining results
from our study revealed that DKK1 was mainly enriched in
functions including cell growth, cell differentiation and tumor
necrosis factor; these functions have been demonstrated to be
correlated with certain cancers (44,45). In
our study, DKK1 was overexpressed in GC cells induced by
TSA. Considering the studies above, one of the possible inhibition
mechanisms of TSA in GC may be that TSA induces histone acetylation
at the DKK1 promoter, which results in the reactivation of
DKK1 expression (37,38). In other words, DKK1 may be a
significant therapeutic target for GC.
KLF13 is a zinc finger transcription factor
known to play a role in proliferation, differentiation, cell cycle
progression and apoptosis (46–48).
KLF13 is required for the expression of several oncogenes
including cyclin D1, which is a known oncogene in oral squamous
cell carcinoma (47,49). Furthermore, KLF13 is believed
to play a role in cancer, and altered expression of it contributes
to tumorigenesis (50–53). Henson et al revealed that
KLF13 was overexpressed in oral cancer cells, and
artificially reducing its cellular levels decreases cell
proliferation and malignancy; therefore, KLF13 may be a
useful biomarker for early detection and a possible target for
therapy (49). KLF13 is also
involved in the proliferation and differentiation of the heart
(47,54). KLF13 was downregulated in our
study. All of the studies above indicated that decreased expression
of KLF13 may be a possible inhibition mechanism of GC
induced by TSA. In summary, KLF13 may be utilized as a
biomarker for detection and a new therapeutic target for GC.
In conclusion, the propanoate metabolism pathway and
the associated genes DKK1 and KLF13 may play
significant roles in the inhibition of GC induced by TSA. They may
be possible therapeutic targets for GC. However, further studies
are necessary to verify the clinical applications of these genes as
biological targets for GC treatment.
References
|
1
|
Parkin DM, Pisani P and Ferlay J:
Estimates of the worldwide incidence of 25 major cancers in 1990.
Int J Cancer. 80:827–841. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neugut AI, Hayek M and Howe G:
Epidemiology of gastric cancer. Semin Oncol. 23:281–291.
1996.PubMed/NCBI
|
|
3
|
Hippo Y, Taniguchi H, Tsutsumi S, Machida
N, Chong JM, Fukayama M, Kodama T and Aburatani H: Global gene
expression analysis of gastric cancer by oligonucleotide
microarrays. Cancer Res. 62:233–240. 2002.PubMed/NCBI
|
|
4
|
Zhang X, Yashiro M, Ren J and Hirakawa K:
Histone deacetylase inhibitor, trichostatin A, increases the
chemosensitivity of anticancer drugs in gastric cancer cell lines.
Oncol Rep. 16:563–568. 2006.PubMed/NCBI
|
|
5
|
Wu ZQ, Zhang R, Chao C, Zhang JF and Zhang
YQ: Histone deacetylase inhibitor trichostatin A induced
caspase-independent apoptosis in human gastric cancer cell. Chin
Med J (Engl). 120:2112–2118. 2007.PubMed/NCBI
|
|
6
|
Lee HS, Park MH, Yang SJ, Jung HY, Byun
SS, Lee DS, Yoo HS, Yeom YI and Seo SB: Gene expression analysis in
human gastric cancer cell line treated with trichostatin A and
S-adenosyl-L-homocysteine using cDNA microarray. Biol Pharm Bull.
27:1497–1503. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zou XM, Li YL, Wang H, Cui W, Li XL, Fu SB
and Jiang HC: Gastric cancer cell lines induced by trichostatin A.
World J Gastroenterol. 14:4810–4815. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Friedgen B, Wölfel R, Russ H, Schömig E
and Graefe KH: The role of extraneuronal amine transport systems
for the removal of extracellular catecholamines in the rabbit.
Naunyn Schmiedebergs Arch Pharmacol. 354:275–286. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen H, Tini M and Evans RM: HATs on and
beyond chromatin. Curr Opin Cell Biol. 13:218–224. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Strahl BD, Briggs SD, Brame CJ, Caldwell
JA, Koh SS, Ma H, Cook RG, Shabanowitz J, Hunt DF, Stallcup MR and
Allis CD: Methylation of histone H4 at arginine 3 occurs in vivo
and is mediated by the nuclear receptor coactivator PRMT1. Curr
Biol. 11:996–1000. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheung WL, Briggs SD and Allis CD:
Acetylation and chromosomal functions. Curr Opin Cell Biol.
12:326–333. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshida M, Kijima M, Akita M and Beppu T:
Potent and specific inhibition of mammalian histone deacetylase
both in vivo and in vitro by trichostatin A. J Biol Chem.
265:17174–17179. 1990.PubMed/NCBI
|
|
13
|
Finnin MS, Donigian JR, Cohen A, Richon
VM, Rifkind RA, Marks PA, Breslow R and Pavletich NP: Structures of
a histone deacetylase homologue bound to the TSA and SAHA
inhibitors. Nature. 401:188–193. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kosugi H, Towatari M, Hatano S, Kitamura
K, Kiyoi H, Kinoshita T, Tanimoto M, Murate T, Kawashima K, Saito H
and Naoe T: Histone deacetylase inhibitors are the potent
inducer/enhancer of differentiation in acute myeloid leukemia: a
new approach to anti-leukemia therapy. Leukemia. 13:1316–1324.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tusher VG, Tibshirani R and Chu G:
Significance analysis of microarrays applied to the ionizing
radiation response. Proc Natl Acad Sci USA. 98:5116–5121. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yekutieli D and Benjamini Y:
Resampling-based false discovery rate controlling multiple test
procedures for correlated test statistics. J Stat Plan Infer.
82:171–196. 1999. View Article : Google Scholar
|
|
17
|
Hulsegge I, Kommadath A and Smits MA:
Globaltest and GOEAST: two different approaches for gene ontology
analysis. BMC Proc. 3:(Suppl 4). S102009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
da W Huang, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
19
|
Ogata H, Goto S, Sato K, Fujibuchi W, Bono
H and Kanehisa M: KEGG: Kyoto encyclopedia of genes and genomes.
Nucleic Acids Res. 27:29–34. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Papadopoulos GL, Reczko M, Simossis VA,
Sethupathy P and Hatzigeorgiou AG: The database of experimentally
supported targets: a functional update of TarBase. Nucleic Acids
Res. 37:(Database issue). D155–D158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garcia DM, Baek D, Shin C, Bell GW,
Grimson A and Bartel DP: Weak seed-pairing stability and high
target-site abundance decrease the proficiency of lsy-6 and other
microRNAs. Nat Struct Mol Biol. 18:1139–1146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: miRecords: an integrated resource for microRNA-target
interactions. Nucleic Acids Res. 37:(Database issue). D105–D110.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: a
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang JH, Zhao LF, Lin P, Su XR, Chen SJ,
Huang LQ, Wang HF, Zhang H, Hu ZF, Yao KT and Huang ZX: GenCLiP
2.0: a web server for functional clustering of genes and
construction of molecular networks based on free terms.
Bioinformatics. 30:2534–2536. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu YP, Bian XJ, Ye DW, Yao XD, Zhang SL,
Dai B, Zhang HL and Shen YJ: Long noncoding RNA expression
signatures of bladder cancer revealed by microarray. Oncol Lett.
7:1197–1202. 2014.PubMed/NCBI
|
|
26
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote cancer
metastasis. Nature. 464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang F, Bi J, Xue X, Zheng L, Zhi K, Hua J
and Fang G: Up-regulated long non-coding RNA H19 contributes to
proliferation of gastric cancer cells. FEBS J. 279:3159–3165. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lai MC, Yang Z, Zhou L, Zhu QQ, Xie HY,
Zhang F, Wu LM, Chen LM and Zheng SS: Long non-coding RNA MALAT-1
overexpression predicts tumor recurrence of hepatocellular
carcinoma after liver transplantation. Med Oncol. 29:1810–1816.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cao DL, Ye DW, Zhang HL, Zhu Y, Wang YX
and Yao XD: A multiplex model of combining gene-based,
protein-based, and metabolite-based with positive and negative
markers in urine for the early diagnosis of prostate cancer.
Prostate. 71:700–710. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang C, Li X, Wang Y, Zhao L and Chen W:
Long non-coding RNA UCA1 regulated cell cycle distribution via CREB
through PI3-K dependent pathway in bladder carcinoma cells. Gene.
496:8–16. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ying L, Chen Q, Wang Y, Zhou Z, Huang Y
and Qiu F: Upregulated MALAT-1 contributes to bladder cancer cell
migration by inducing epithelial-to-mesenchymal transition. Mol
Biosyst. 8:2289–2294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu Y, Yu M and Li Z, Kong C, Bi J, Li J,
Gao Z and Li Z: ncRAN, a newly identified long noncoding RNA,
enhances human bladder tumor growth, invasion, and survival.
Urology. 77(510): e1–e5. 2011.PubMed/NCBI
|
|
33
|
Perroud B, Lee J, Valkova N, Dhirapong A,
Lin PY, Fiehn O, Kültz D and Weiss RH: Pathway analysis of kidney
cancer using proteomics and metabolic profiling. Mol Cancer.
5:642006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gmeiner WH, Hellmann GM and Shen P:
Tissue-dependent and independent gene expression changes in
metastatic colon cancer. Oncol Rep. 19:245–251. 2008.PubMed/NCBI
|
|
35
|
Yang Q, Shen SS, Zhou S, Ni J, Chern D,
Wang G and Li Y: STAT3 activation and aberrant ligand-dependent
sonic hedgehog signaling in human pulmonary adenocarcinoma. Exp Mol
Pathol. 93:227–236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gonzalez-Angulo AM, Iwamoto T, Liu S, Chen
H, Do KA, Hortobagyi GN, Mills GB, Meric-Bernstam F, Symmans WF and
Pusztai L: Gene expression, molecular class changes, and pathway
analysis after neoadjuvant systemic therapy for breast cancer. Clin
Cancer Res. 18:1109–1119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hirata H, Hinoda Y, Nakajima K, Kawamoto
K, Kikuno N, Ueno K, Yamamura S, Zaman MS, Khatri G, Chen Y, et al:
Wnt antagonist DKK1 acts as a tumor suppressor gene that induces
apoptosis and inhibits proliferation in human renal cell carcinoma.
Int J Cancer. 128:1793–1803. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee J, Yoon YS and Chung JH: Epigenetic
silencing of the WNT antagonist DICKKOPF-1 in cervical cancer cell
lines. Gynecol Oncol. 109:270–274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sato H, Suzuki H, Toyota M, Nojima M,
Maruyama R, Sasaki S, Takagi H, Sogabe Y, Sasaki Y, Idogawa M, et
al: Frequent epigenetic inactivation of DICKKOPF family genes in
human gastrointestinal tumors. Carcinogenesis. 28:2459–2466. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang H, Li Q and Chen H: Genistein affects
histone modifications on Dickkopf-related protein 1 (DKK1) gene in
SW480 human colon cancer cell line. PLoS One. 7:e409552012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yamabuki T, Takano A, Hayama S, Ishikawa
N, Kato T, Miyamoto M, Ito T, Ito H, Miyagi Y, Nakayama H, et al:
Dikkopf-1 as a novel serologic and prognostic biomarker for lung
and esophageal carcinomas. Cancer Res. 67:2517–2525. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sheng SL, Huang G, Yu B and Qin WX:
Clinical significance and prognostic value of serum Dickkopf-1
concentrations in patients with lung cancer. Clin Chem.
55:1656–1664. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Park H, Jung HY, Choi HJ, Kim DY, Yoo JY,
Yun CO, Min JK, Kim YM and Kwon YG: Distinct roles of DKK1 and DKK2
in tumor angiogenesis. Angiogenesis. 17:221–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Katzenellenbogen BS: Estrogen receptors:
bioactivities and interactions with cell signaling pathways. Biol
Reprod. 54:287–293. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Katzenellenbogen BS, Montano MM, Ekena K,
Herman ME and McInerney EM: William L. McGuire memorial lecture.
Antiestrogens: mechanisms of action and resistance in breast
cancer. Breast Cancer Res Treat. 44:23–38. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen X, Johns DC, Geiman DE, Marban E,
Dang DT, Hamlin G, Sun R and Yang VW: Krüppel-like factor 4
(gut-enriched Krüppel-like factor) inhibits cell proliferation by
blocking G1/S progression of the cell cycle. J Biol Chem.
276:30423–30428. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nemer M and Horb ME: The KLF family of
transcriptional regulators in cardiomyocyte proliferation and
differentiation. Cell Cycle. 6:117–121. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kaczynski J, Cook T and Urrutia R: Sp1-
and Krüppel-like transcription factors. Genome Biol. 4:2062003.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Henson BJ and Gollin SM: Overexpression of
KLF13 and FGFR3 in oral cancer cells. Cytogenet Genome Res.
128:192–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Black AR, Black JD and Azizkhan-Clifford
J: Sp1 and krüppel-like factor family of transcription factors in
cell growth regulation and cancer. J Cell Physiol. 188:143–160.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dang DT, Mahatan CS, Dang LH, Agboola IA
and Yang VW: Expression of the gut-enriched Krüppel-like factor
(Krüppel-like factor 4) gene in the human colon cancer cell line
RKO is dependent on CDX2. Oncogene. 20:4884–4890. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Miller KA, Eklund EA, Peddinghaus ML, Cao
Z, Fernandes N, Turk PW, Thimmapaya B and Weitzman SA: Kruppel-like
factor 4 regulates laminin alpha 3A expression in mammary
epithelial cells. J Biol Chem. 276:42863–42868. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Martin KM, Ellis PD, Metcalfe JC and Kemp
PR: Selective modulation of the SM22alpha promoter by the binding
of BTEB3 (basal transcription element-binding protein 3) to TGGG
repeats. Biochem J. 375:457–463. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lavallée G, Andelfinger G, Nadeau M,
Lefebvre C, Nemer G, Horb ME and Nemer M: The Kruppel-like
transcription factor KLF13 is a novel regulator of heart
development. EMBO J. 25:5201–5213. 2006. View Article : Google Scholar : PubMed/NCBI
|