Introduction
Pancreatic cancer is relatively rare, with an
incidence of ~12 cases per 100,000 people in the USA and 2–8 cases
per 100,000 people worldwide (1,2). However,
pancreatic cancer is the fourth-leading cause of cancer mortalities
in men and women in the USA, and the eighth- and ninth- leading
cause of cancer mortalities in men and women worldwide,
respectively (2,3). Pancreatic ductal adenocarcinoma (PDAC)
accounts for 85% of all pancreatic cancer cases (4). The disease tends to occur in the elderly
and present at an advanced stage, with a median age at diagnosis of
71 years old, and <20% of cases presenting with localized and
resectable tumors (4,5). Advances in treatment, specifically
multi-agent chemotherapy, have improved survival rates, which were
previously poor in those with advanced and metastatic pancreatic
cancer (4). In addition, adjuvant
therapy has been shown to confer a survival advantage compared with
postoperative observation alone in resected pancreatic cancer
(4–6).
However, despite these advances, pancreatic cancer remains among
the most lethal cancers, with a 5-year survival rate of 6–7% across
all stages (1).
In an effort to improve outcomes via disease
prevention, a growing body of research has focused on the
environmental risk factors for pancreatic cancer (7–9). The
association between pancreatic cancer and modifiable risk factors
with moderate/strong evidence of association, including tobacco
use, alcoholism and dietary intake, has been extensively reviewed
(8,9).
It has been estimated that ~2/3 of the major risk factors for
pancreatic cancer are potentially modifiable (9). Furthermore, there is evidence to suggest
that 1/3 of the burden of pancreatic cancer may be prevented
through the application of this knowledge (9). Evidence for the chemopreventive effects
of medications typically used to treat non-cancer-related and
chronic conditions is increasing (10). In particular, the potential
application of statins as a chemopreventive agent for pancreatic
cancer is an area of increasing interest and is evaluated in the
present review.
Metabolic syndrome and pancreatic cancer
risk
The presence of metabolic syndrome, defined in one
meta-analysis as the co-occurrence of ≥3 medical conditions,
including diabetes, hypertension, hyperlipidemia, and a body mass
index (BMI) ≥25 kg/m2, has been shown to be associated
with a relative risk (RR) for pancreatic cancer of 1.55 [95%
confidence interval (CI), 1.19–2.01], with a greater risk for those
patients with more components of the syndrome (11). In addition, meta-analyses have shown a
stronger association between pancreatic cancer and metabolic
syndrome in women (11–13). Furthermore, individual components of
metabolic syndrome have been identified as modifiable risk factors
for pancreatic cancer. For example, diabetes as a risk factor for
the development of pancreatic cancer, leading to the assessment of
metformin as a potential chemopreventive agent for this cancer
(14).
Several pooled analyses and meta-analyses have
demonstrated that obesity is a risk factor for pancreatic cancer,
with a RR of <1.47 (95% CI, 1.23–1.75) in those with a BMI ≥30
kg/m2, and which is often independent of other risk
factors, such as age, gender, diabetes and smoking status (8,9,15–21). The
RR for pancreatic cancer following an increase of 5 units in BMI,
10 cm in waist circumference and 0.1 unit in waist-to-hip ratio has
been shown to be 1.10 (95% CI, 1.07–1.14), 1.11 (95% CI, 1.05–1.18)
and 1.19 (95% CI, 1.09–1.31), respectively (20). Compared with individuals who were not
overweight (BMI <25 kg/m2) or obese (BMI <30
kg/m2) in early adulthood (defined as age 18 or 21
years), the risk for pancreatic cancer was higher in those who were
overweight or obese in early adulthood (RR, 1.54; 95% CI,
1.24–1.93) (19). Furthermore,
pancreatic cancer risk was greater (RR, 1.40; 95% CI, 1.13–1.72) in
those who gained ≥10 kg/m2 in BMI at baseline and early
adulthood compared with those whose BMI remained stable (19). However, BMI does not appear to be
associated with risk of mortality from pancreatic cancer in the
Asian population, following adjustment for risk factors, including
age, smoking status and diabetes (22).
Fatty infiltration of the pancreas, which is
determined histopathologically, appears to be a risk factor for
pancreatic intraepithelial neoplasias (PanINs) and PDAC, with BMI
being the most significantly associated factor with pancreatic
fatty infiltration (23,24). Notably, circulating levels of
adiponectin, an adipocyte-secreted hormone with insulin sensitizing
and anti-inflammatory properties, has been revealed to be inversely
correlated with pancreatic cancer risk, independent of other risk
factors (25). Notably, meta-analyses
of other anthropometric measures such as physical activity have
primarily produced non-significant findings in relation to
protective effects against pancreatic cancer (26–28).
Consistent physical activity over time was demonstrated to have a
greater association with pancreatic cancer risk reduction (RR,
0.86; 95% CI, 0.76–0.97) compared with recent (RR, 0.95; 95% CI,
0.90–1.01) and distant (RR, 0.95; 95% CI, 0.79–1.15) past physical
activity alone (28).
Abnormal lipid metabolism, which can cause high
levels of total cholesterol and triglycerides, or low levels of
high-density lipoprotein and apolipoprotein A-I, has been
identified to be associated with an increased risk of
obesity-related cancers, including pancreatic cancer, in a
meta-analysis (29). A separate
pooled analysis and meta-analysis demonstrated that total serum
cholesterol is inversely correlated with pancreatic cancer risk in
men (30) and the European population
(31). In addition, meta-analyses
have shown that a high dietary intake of cholesterol is
significantly associated with risk for pancreatic cancer,
particularly in the American and European populations, with RRs
<1.371 (95% CI, 1.155–1.627), following adjustment for
confounding factors (31,32). Furthermore, pancreatic cancer risk
rises by 8% with an increased intake in cholesterol of 100 mg/day
(RR, 1.08; 95% CI, 1.04–1.13), in what was determined to a linear
dose-response association (32).
Total dietary fat consumption was reported to be independently
associated with pancreatic cancer risk according to one
meta-analysis (33), although this
has been refuted (8). Results
regarding the association between omega-3 polyunsaturated fatty
acid consumption and pancreatic cancer risk/survival are mixed
(34,35).
Statins and pancreatic cancer
Epidemiologic evidence for the association between
statins and pancreatic cancer risk remains controversial. Several
epidemiologic studies, including two meta-analyses, have suggested
that statin use, regardless of dose, duration or type (lipophilic,
such as atorvastatin, lovastatin and simvastatin, or hydrophilic,
such as pravastatin, rosuvastatin and fluvastatin), does not
significantly affect pancreatic cancer risk (36–40).
However, significant heterogeneity was detected among the included
studies (36,40). Conversely, a dose-response association
was identified between statin use and pancreatic cancer risk with
an 80% reduced pancreatic cancer risk [odds ratio (OR), 0.20; 95%
CI, 0.13–0.29] in those with >4 years statin use, irrespective
of age, gender, ethnicity, BMI, diabetes, smoking status or alcohol
use (41). Furthermore, in a matched
case-control study, male smokers who were regular statin users had
a significantly reduced OR for pancreatic cancer (42). In a large case-control study (1,405
participants), statin use in men (OR, 0.50; 95% CI, 0.32–0.79) and
a duration of statin use of >10 years (OR, 0.51; 95% CI,
0.31–0.85) were significantly associated with a reduced risk of
pancreatic cancer (43).
Notably, preoperative use of statins has been
demonstrated to be a predictor of increased early postoperative
mortality in patients with resected pancreatic cancer (44). However, one study reported that
statins improved the survival [hazard ratio (HR), 0.40; P=0.010] of
patients with diabetes who were undergoing chemotherapy for
advanced pancreatic cancer (45).
Furthermore, patients with metabolic syndrome and pancreatic cancer
on hydrophilic statins exhibited increased survival (HR, 0.67; 95%
CI, 0.46–0.96) compared with those not on statins (46).
Mechanisms of action and evidence of
antitumor effects of statins in pancreatic cancer
Early evidence demonstrated that lovastatin and
simvastatin, inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A
(HMG-CoA) reductase, the rate-limiting enzyme in the
mevalonate/cholesterol synthesis pathway, inhibited tumor growth in
a number of human and rodent pancreatic cancer cell lines in
vitro and tumor xenografts in mice in vivo, through cell
cycle arrest in G1 and inhibition of DNA synthesis, and that this
process was reversed by adding mevalonic acid (47–49). In
addition, lovastatin was noticed to induce G1 and G2/M cell cycle
arrest, thus inhibiting tumor growth in vitro in a
dose-dependent manner, which was possibly independent of K-Ras
mutational status, and to significantly decrease post-translational
phosphorylation of the Ras protein in K-Ras mutant human pancreatic
cancer cells (50). Furthermore, the
results of another study indicated that statins exerted direct
cytotoxic effects in human pancreatic cancer cells via
pro-apoptotic activity (51).
Fluvastatin was identified to inhibit epidermal
growth factor (EGF)-induced invasion of human pancreatic cancer
cells in vitro, and to reduce metastatic tumor formation and
growth in vivo at the dose recommended for the treatment of
hypercholesterolemia in humans (52,53).
Specifically, fluvastatin prevents geranylgeranylation of RhoA, a
member of the Rho subfamily of small guanosine triphosphatases that
are involved in cell motility, structure and invasion (52,53).
Geranylgeranylation is a form of post-translational modification
known as prenylation, by which isoprene units from an isoprenoid
intermediate of the mevalonate/cholesterol synthesis pathway such
as geranylgeranyl pyrophosphate are attached to target proteins
(52,53). Inhibition of geranylgeranylation of
RhoA inhibits EGF-induced translocation of RhoA from the cytoplasm
to the plasma membrane and its subsequent activation (52,53). In a
separate study, fluvastatin was shown to inhibit farnesylation [the
attachment of a lipophilic farnesyl group from farnesyl
pyrophosphate (FPP) to a cysteine residue at the C-terminus of a
target protein] of Ras in a dose-dependent manner, a process
critical for the membrane translocation and subsequent activation
of Ras, which is involved in cell growth, proliferation and
survival (Fig. 1) (54). In addition, fluvastatin reduces
phosphorylation and activation of mitogen-activated protein kinase
1 (MAPK1)/extracellular signal-regulated kinase 2 (ERK2), a
downstream effector of Ras, and synergizes with gemcitabine to
significantly inhibit tumor growth in vitro and in
vivo through increasing the expression of an enzyme required
for the activation of gemcitabine (54).
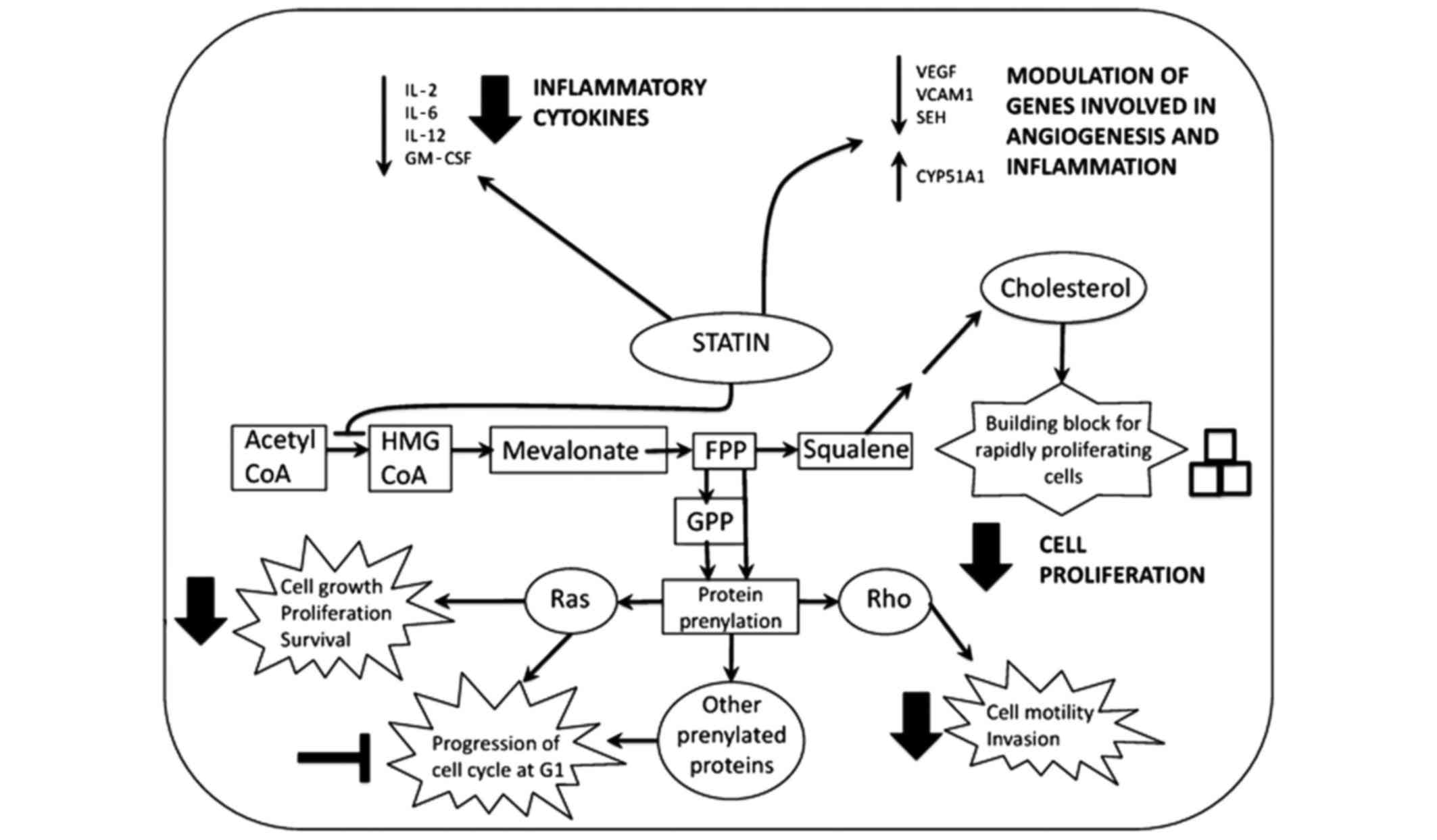 | Figure 1.Mechanism of action of statins in
pancreatic cancer. Statins decrease the expression of inflammatory
cytokines and modulate the expression of a number of genes involved
in angiogenesis and inflammation, which may protect against
carcinogenesis. In addition, statins inhibit protein prenylation.
This prevents the proper functioning of guanosine triphosphatase
proteins such as Ras and Rho, thus inhibiting downstream pathways
that are involved in cell growth, proliferation, survival, motility
and invasion, which leads to cell cycle arrest in G1. Furthermore,
statins impair cancer cell proliferation by inhibiting the
synthesis of cholesterol, which is essential for new membrane
formation in rapidly proliferating cells. IL, interleukin; GM-CSF,
granulocyte macrophage colony-stimulating factor; VEGF, vascular
endothelial growth factor; VCAM1, vascular cell adhesion molecule
1; SEH, soluble epoxide hydrolase; CYP51A1, cytochrome P450 family
51 subfamily A member 1; Acetyl-CoA, acetyl-coenzyme A; HMG-CoA,
3-hydroxy-3-methylglutaryl-coenzyme A; FPP, farnesyl pyrophosphate;
GPP, geranyl pyrophosphate. |
The importance of isoprenoid synthesis and
prenylation in pancreatic cancer growth was reinforced when
lovastatin in combination with pamidronate, an inhibitor of FPP
synthase, was demonstrated to inhibit farnesylation and
geranylgeranylation, and to exhibit synergistic antitumor effects
in vitro and in vivo (55). Furthermore, statins were shown to
improve survival and reduce tumor burden in animal models of
pancreatic cancer compared with controls at doses below the maximum
recommended dose for humans (56).
Tumor tissues from K-Ras mutant mice treated with atorvastatin
displayed significantly decreased levels of membrane-bound K-Ras
and phosphorylated Raf (56). In
addition, an analysis of pancreatic cancer cell lines treated with
atorvastatin in vitro demonstrated a dose-dependent
reduction in the activation of downstream effectors of Ras,
including Raf, ERK1/2, Jun and p90 ribosomal s6 kinase, and an
inhibition of targets of prenylation that are important for
protein-protein interactions and carcinogenesis, such as human DnaJ
homolog and nuclear prelamin A (56).
Of note, in vitro gene array analysis revealed that
atorvastatin modulated the expression of 132 genes, including those
involved in inflammation, such as cytochrome P450 family 51
subfamily A member 1, soluble epoxide hydrolase and vascular
adhesion molecule 1 (Fig. 1)
(56).
Simvastatin and atorvastatin have been shown to
significantly delay the progression of PanIN lesions to PDAC and to
inhibit PDAC growth in conditional K-Ras mutant mice (57,58). In
one study, treatment with atorvastatin resulted in significantly
downregulated expression of components of the Ras/MAPK,
phosphoinositide 3-kinase (PI3K)/Akt and nuclear factor-κB (NF-κB)
signaling pathways, including Akt, phosphorylated (p) Akt,
purinergic receptor P2X7, RhoA, pERK, cyclin dependent kinase 2,
cyclin D1, β-catenin, cyclin E, survivin, caveolin-1,
granulocyte-macrophage colony-stimulating factor, cyclooxygenase-2
(COX-2), and interleukin (IL)-2, −6 and −12 in vivo
(Fig. 1) (58). The ability of atorvastatin to inhibit
PI3K/Akt, Ras/MAPK and NF-κB signaling may be dependent on the
underexpression of P2X7, at least in human pancreatic cancer cells
in vitro (59). Combined
inhibition of HMG-CoA reductase, COX-2 and farnesyl transferase in
human pancreatic cancer cell lines in vitro was observed to
inhibit tumor growth and to produce a stronger decrease in pAkt and
pERK1/2 levels compared with each agent alone (60). A summary of the antitumor mechanisms
of statins in pancreatic cancer from existing evidence is shown in
Fig. 1.
The association between statins, lipid metabolism
and PDAC development continues to grow in complexity. A previous
study identified a potential role for simvastatin-induced
accumulation of cytosolic lipid droplets and upregulation of genes
involved in lipid metabolism [such as ATP-binding cassette (ABC)
A7] and triacylglycerole and phospholipid synthesis (such as
1-acylglycerol-3-phosphate O-acyltransferase 2) in the apoptosis of
pancreatic cancer cells in vitro (61). Oxysterol binding protein-like 5 (ORP5)
has been associated with increased pancreatic cancer invasion,
stimulation of cholesterol synthesis via expression of sterol
response element binding protein 2 (SREBP2) and regulation of the
effects of statins in human PDAC cells in vitro (62). High doses of statins were required for
growth inhibition in cancer cells strongly expressing ORP5, while
only low doses were required in cells with moderate expression
(62). In addition, a growing body of
research has identified that targeting mediators of
mevalonate/cholesterol synthesis and lipid metabolism other than
HMG-CoA reductase with statins or alternative inhibitors modulates
chemotherapy and radiation resistance in pancreatic cancer, and
suppresses pancreatic cancer growth and metastasis (63–67).
A recent preclinical study highlighted that the
majority of activated pro-tumorigenic metabolic pathways in K-Ras
mutant mouse models of PDAC were involved in lipid metabolism
compared with control mice (68). In
particular, the expression of low-density lipoprotein receptor
(LDLR), apolipoprotein (Apo) B, ApoE, ABCA1, SREBP2, HMG-CoA
reductase, cholesterol acyltransferase 1, lipase A, and other
transcripts involved in the synthesis/uptake of cholesterol and its
derivatives (including oxysterols and steroid hormones) was
significantly increased in PDAC compared with control pancreas in
mouse models in vivo, illustrating the high dependence of
pancreatic cancer cells on cholesterol (68). In addition, an increase in total
cholesterol, cholesterol ester (CE) and free cholesterol (FC)
content was observed in pancreatic tumors compared with healthy
pancreas (68). Knockdown of LDLR
expression in vitro via short hairpin RNA was demonstrated
to inhibit cell proliferation, enhance the cytotoxic effects of
chemotherapy and redistribute total cholesterol content (reduce CE
and increase FC) in PDAC, without compensatory overactivation of
cholesterol synthesis (68).
Furthermore, enhanced LDLR expression appears to be an indicator of
poor prognosis in human PDAC, regardless of cancer stage (68).
The existence of differential effects between
hydrophobic statins such as simvastatin and hydrophilic statins
such as pravastatin have been proposed, as simvastatin appears to
produce greater antitumor effects compared with pravastatin in
several cancer cell lines (excluding pancreatic) in vitro
(69). However, another study
demonstrated that, although simvastatin produced the greatest
antitumor effects in vitro, rosuvastatin, cerivastatin and
fluvastatin had the most potent antitumor effects in animal models
of PDAC at the recommended human doses, with all statins
(hydrophobic and hydrophilic) except pravastatin exerting
inhibitory effects on intracellular Ras translocation (70). Conversely, a phase II trial involving
114 patients with locally advanced and metastatic pancreatic cancer
revealed that patients who received 3 weeks of gemcitabine (1,000
mg/m2 on days 1, 8 and 15) and simvastatin (40 mg once
daily) had no clinical benefit compared with patients treated with
gemcitabine alone, although there was no increase in toxicity from
the combined treatment (71).
Conclusions
Although statins in combination with chemotherapy
have failed to demonstrate improved antitumor efficacy based on
recent clinical evidence, the data are limited to just a few
clinical trials in patients with advanced and metastatic PDAC
(71). The attractiveness of statins
as a component of chemotherapeutic regimens for pancreatic cancer
lies in their relatively safe and well-tolerated toxicity profile
and low cost (10). For this reason,
further clinical trials are warranted to better define the
potential of statins as an adjunct to standard chemotherapy for the
treatment of pancreatic cancer. Aside from treatment, disease
prevention represents another opportunity to improve patient
outcomes in pancreatic cancer. Metabolic syndrome and its
components, including diabetes, obesity and dyslipidemia, have been
recognized as modifiable risk factors for pancreatic cancer
(72,73). Evidence discussed in the present
review has demonstrated that statins potentially reduce pancreatic
cancer risk and improve survival in patients with a combination of
metabolic syndrome and pancreatic cancer. Furthermore, preclinical
studies suggest that statins exhibit antitumor effects in
pancreatic cancer cell lines in vitro and in animal models
in vivo, in addition to delaying the progression of PanIN
lesions to PDAC and inhibiting PDAC formation in conditional K-Ras
mice models. However, the mechanisms by which statins elicit these
effects remain poorly understood, although recent evidence
postulates that statins inhibit the mevalonate/cholesterol
synthesis pathway, thus blocking the formation of intermediates
that are critical for prenylation and activation of Ras/MAPK,
PI3K/Akt and associated signaling pathways (10). In addition, statins appear to modulate
lipid metabolism and inflammation signaling pathways, which are
important for pancreatic cancer growth and progression (73). In conclusion, it is evident that
further studies are required to elucidate the mechanisms underlying
the anticancer effects of statins in PDAC. Nevertheless, increasing
evidence supports the potential application of statins as a
chemopreventive agent in pancreatic cancer.
References
|
1
|
Howlader N, Noone AM, Krapcho M, Garshell
J, Neyman N, Altekruse SF, et al: SEER Cancer Statistics Review,
1975–2011. National Cancer Institute; Bethesda, MD: http://seer.cancer.gov/csr/1975_2011/2014
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ryan DP, Hong TS and Bardeesy N:
Pancreatic adenocarcinoma. N Engl J Med. 371:1039–1049. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wolfgang CL, Herman JM, Laheru DA, Klein
AP, Erdek MA, Fishman EK and Hruban RH: Recent progress in
pancreatic cancer. CA Cancer J Clin. 63:318–348. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lowenfels AB and Maisonneuve P: Can we
prevent pancreatic disease? Clin Gastroenterol Hepatol.
12:1645–1646. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alsamarrai A, Das SL, Windsor JA and
Petrov MS: Factors that affect risk for pancreatic disease in the
general population: A systematic review and meta-analysis of
prospective cohort studies. Clin Gastroenterol Hepatol.
12:1635.e5–1644.e5; quiz e103. 2014. View Article : Google Scholar
|
|
9
|
Maisonneuve P and Lowenfels AB: Risk
factors for pancreatic cancer: A summary review of meta-analytical
studies. Int J Epidemiol. 44:186–198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gronich N and Rennert G: Beyond
aspirin-cancer prevention with statins, metformin and
bisphosphonates. Nat Rev Clin Oncol. 10:625–642. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rosato V, Tavani A, Bosetti C, Pelucchi C,
Talamini R, Polesel J, Serraino D, Negri E and La Vecchia C:
Metabolic syndrome and pancreatic cancer risk: A case-control study
in Italy and meta-analysis. Metabolism. 60:1372–1378. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Esposito K, Chiodini P, Colao A, Lenzi A
and Giugliano D: Metabolic syndrome and risk of cancer: A
systematic review and meta-analysis. Diabetes Care. 35:2402–2411.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stocks T, Bjørge T, Ulmer H, Manjer J,
Häggström C, Nagel G, Engeland A, Johansen D, Hallmans G, Selmer R,
et al: Metabolic risk score and cancer risk: Pooled analysis of
seven cohorts. Int J Epidemiol. 44:1353–1363. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gong J, Robbins LA, Lugea A, Waldron RT,
Jeon CY and Pandol SJ: Diabetes, pancreatic cancer, and metformin
therapy. Front Physiol. 5:4262014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
de Gonzalez A Berrington, Sweetland S and
Spencer E: A meta-analysis of obesity and the risk of pancreatic
cancer. Br J Cancer. 89:519–523. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Larsson SC, Orsini N and Wolk A: Body mass
index and pancreatic cancer risk: A meta-analysis of prospective
studies. Int J Cancer. 120:1993–1998. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arslan AA, Helzlsouer KJ, Kooperberg C,
Shu XO, Steplowski E, Bueno-de-Mesquita HB, Fuchs CS, Gross MD,
Jacobs EJ, Lacroix AZ, et al: Anthropometric measures, body mass
index and pancreatic cancer: A pooled analysis from the Pancreatic
Cancer Cohort Consortium (PanScan). Arch Intern Med. 170:791–802.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiao L, de Gonzalez A Berrington, Hartge
P, Pfeiffer RM, Park Y, Freedman DM, Gail MH, Alavanja MC, Albanes
D, Freeman LE Beane, et al: Body mass index, effect modifiers, and
risk of pancreatic cancer: A pooled study of seven prospective
cohorts. Cancer Causes Control. 21:1305–1314. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Genkinger JM, Spiegelman D, Anderson KE,
Bernstein L, van den Brandt PA, Calle EE, English DR, Folsom AR,
Freudenheim JL, Fuchs CS, et al: A pooled analysis of 14 cohort
studies of anthropometric factors and pancreatic cancer risk. Int J
Cancer. 129:1708–1717. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aune D, Greenwood DC, Chan DS, Vieira R,
Vieira AR, Rosenblatt DA Navarro, Cade JE, Burley VJ and Norat T:
Body mass index, abdominal fatness and pancreatic cancer risk: A
systematic review and non-linear dose-response meta-analysis of
prospective studies. Ann Oncol. 23:843–852. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bethea TN, Kitahara CM, Sonderman J, Patel
AV, Harvey C, Knutsen SF, Park Y, Park SY, Fraser GE, Jacobs EJ, et
al: A pooled analysis of body mass index and pancreatic cancer
mortality in African Americans. Cancer Epidemiol Biomarkers Prev.
23:2119–2125. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin Y, Fu R, Grant E, Chen Y, Lee JE,
Gupta PC, Ramadas K, Inoue M, Tsugane S, Gao YT, et al: Association
of body mass index and risk of death from pancreatic cancer in
Asians: Findings from the Asia Cohort Consortium. Eur J Cancer
Prev. 22:224–250. 2013. View Article : Google Scholar
|
|
23
|
Hori M, Takahashi M, Hiraoka N, Yamaji T,
Mutoh M, Ishigamori R, Furuta K, Okusaka T, Shimada K, Kosuge T, et
al: Association of pancreatic fatty infiltration with pancreatic
ductal adenocarcinoma. Clin Transl Gastroenterol. 5:e532014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rebours V, Gaujoux S, d'Assignies G,
Sauvanet A, Ruszniewski P, Levy P, Bedossa P, Paradis V and
Couvelard A: Obesity and fatty pancreatic infiltration are risk
factors for pancreatic precancerous lesions (PanIN). Clin Cancer
Res. 21:3522–3528. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bao Y, Giovannucci EL, Kraft P, Stampfer
MJ, Ogino S, Ma J, Buring JE, Sesso HD, Lee IM, Gaziano JM, et al:
A prospective study of plasma adiponectin and pancreatic cancer
risk in five US cohorts. J Natl Cancer Inst. 105:95–103. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bao Y and Michaud DS: Physical activity
and pancreatic cancer risk: A systematic review. Cancer Epidemiol
Biomarkers Prev. 17:2671–2682. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
O'Rorke MA, Cantwell MM, Cardwell CR,
Mulholland HG and Murray LJ: Can physical activity modulate
pancreatic cancer risk? a systematic review and meta-analysis. Int
J Cancer. 126:2957–2968. 2010.PubMed/NCBI
|
|
28
|
Behrens G, Jochem C, Schmid D, Keimling M,
Ricci C and Leitzmann MF: Physical activity and risk of pancreatic
cancer: A systematic review and meta-analysis. Eur J Epidemiol.
30:279–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Melvin JC, Holmberg L, Rohrmann S, Loda M
and Van Hemelrijck M: Serum lipid profiles and cancer risk in the
context of obesity: Four meta-analyses. J Cancer Epidemiol.
2013:8238492013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Strohmaier S, Edlinger M, Manjer J, Stocks
T, Bjørge T, Borena W, Häggström C, Engeland A, Nagel G, Almquist
M, et al: Total serum cholesterol and cancer incidence in the
Metabolic syndrome and Cancer Project (Me-Can). PLoS One.
8:e542422013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen X, Zhou T and Chen M: Meta analysis
of the association of cholesterol with pancreatic carcinoma risk. J
BUON. 20:109–113. 2014. View Article : Google Scholar
|
|
32
|
Chen H, Qin S, Wang M, Zhang T and Zhang
S: Association between cholesterol intake and pancreatic cancer
risk: Evidence from a meta-analysis. Sci Rep. 5:82432015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen QW and Yao QY: Total fat consumption
and pancreatic cancer risk: A meta-analysis of epidemiologic
studies. Eur J Cancer Prev. 24:278–285. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qin B, Xun P and He K: Fish or long-chain
(n-3) PUFA intake is not associated with pancreatic cancer risk in
a meta-analysis and systematic review. J Nutr. 142:1067–1073. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma YJ, Yu J, Xiao J and Cao BW: The
consumption of omega-3 polyunsaturated fatty acids improves
clinical outcomes and prognosis in pancreatic cancer patients: A
systematic evaluation. Nutr Cancer. 67:112–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bonovas S, Filioussi K and Sitaras NM:
Statins are not associated with a reduced risk of pancreatic cancer
at the population level, when taken at low doses for managing
hypercholesterolemia: Evidence from a meta-analysis of 12 studies.
Am J Gastroenterol. 103:2646–2651. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bradley MC, Hughes CM, Cantwell MM and
Murray LJ: Statins and pancreatic cancer risk: A nested
case-control study. Cancer Causes Control. 21:2093–2100. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chiu HF, Chang CC, Ho SC, Wu TN and Yang
CY: Statin use and the risk of pancreatic cancer: A
population-based case-control study. Pancreas. 40:669–672. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jacobs EJ, Newton CC, Thun MJ and Gapstur
SM: Long-term use of cholesterol-lowering drugs and cancer
incidence in a large United States cohort. Cancer Res.
71:1763–1771. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cui X, Xie Y, Chen M, Li J, Liao X, Shen
J, Shi M, Li W, Zheng H and Jiang B: Statin use and risk of
pancreatic cancer: A meta-analysis. Cancer Causes Control.
23:1099–1111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Khurana V, Sheth A, Caldito G and Barkin
JS: Statins reduce the risk of pancreatic cancer in humans: A
case-control study of half a million veterans. Pancreas.
34:260–265. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Carey FJ, Little MW, Pugh TF, Ndokera R,
Ing H, Clark A, Dennison A, Metcalfe MS, Robinson RJ and Hart AR:
The differential effects of statins on the risk of developing
pancreatic cancer: A case-control study in two centres in the
United Kingdom. Dig Dis Sci. 58:3308–3312. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Walker EJ, Ko AH, Holly EA and Bracci PM:
Statin use and risk of pancreatic cancer: Results from a large,
clinic-based case-control study. Cancer. 121:1287–1294. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chagpar RB, Martin RC, Ahmad SA, Kim HJ,
Rupp C, Weber S, Ebelhar A, Gilbert J, Brinkman A, Winslow E, et
al: Medically managed hypercholesterolemia and insulin-dependent
diabetes mellitus preoperatively predicts poor survival after
surgery for pancreatic cancer. J Gastrointest Surg. 15:551–557.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nakai Y, Isayama H, Sasaki T, Mizuno S,
Sasahira N, Kogure H, Kawakubo K, Yamamoto N, Hirano K, Ijichi H,
et al: Clinical outcomes of chemotherapy for diabetic and
nondiabetic patients with pancreatic cancer: Better prognosis with
statin use in diabetic patients. Pancreas. 42:202–208. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jeon CY, Pandol SJ and Goodman MT:
Survival time in pancreatic cancer patients with metabolic syndrome
varies by use of insulin and statins. Cancer Res. 74:(Suppl 19).
21732014. View Article : Google Scholar
|
|
47
|
Mikulski SM, Viera A, Darzynkiewicz Z and
Shogen K: Synergism between a novel amphibian oocyte ribonuclease
and lovastatin in inducing cytostatic and cytotoxic effects in
human lung and pancreatic carcinoma cell lines. Br J Cancer.
66:304–310. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sumi S, Beauchamp RD, Townsend CM Jr,
Uchida T, Murakami M, Rajaraman S, Ishizuka J and Thompson JC:
Inhibition of pancreatic adenocarcinoma cell growth by lovastatin.
Gastroenterology. 103:982–989. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ura H, Obara T, Nishino N, Tanno S,
Okamura K and Namiki M: Cytotoxicity of simvastatin to pancreatic
adenocarcinoma cells containing mutant ras gene. Jpn J Cancer Res.
85:633–638. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Müller C, Bockhorn AG, Klusmeier S, Kiehl
M, Roeder C, Kalthoff H and Koch OM: Lovastatin inhibits
proliferation of pancreatic cancer cell lines with mutant as well
as with wild-type K-ras oncogene but has different effects on
protein phosphorylation and induction of apoptosis. Int J Oncol.
12:717–723. 1998.PubMed/NCBI
|
|
51
|
Kamiński R, Kozar K, Kopeć M, Basak G,
Skierski JS, Koronkiewicz M, Jakóbisiak M and Gołab J: Discussion
on 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors
reduce human pancreatic cancer cell invasion and metastasis.
Gastroenterology. 123:17472002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kusama T, Mukai M, Iwasaki T, Tatsuta M,
Matsumoto Y, Akedo H and Nakamura H: Inhibition of epidermal growth
factor-induced RhoA translocation and invasion of human pancreatic
cancer cells by 3-hydroxy-3-methylglutaryl-coenzyme a reductase
inhibitors. Cancer Res. 61:4885–4891. 2001.PubMed/NCBI
|
|
53
|
Kusama T, Mukai M, Iwasaki T, Tatsuta M,
Matsumoto Y, Akedo H, Inoue M and Nakamura H:
3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors reduce
human pancreatic cancer cell invasion and metastasis.
Gastroenterology. 122:308–317. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bocci G, Fioravanti A, Orlandi P,
Bernardini N, Collecchi P, Del Tacca M and Danesi R: Fluvastatin
synergistically enhances the antiproliferative effect of
gemcitabine in human pancreatic cancer MIAPaCa-2 cells. Br J
Cancer. 93:319–330. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Issat T, Nowis D, Legat M, Makowski M,
Klejman MP, Urbanski J, Skierski J, Koronkiewicz M, Stoklosa T,
Brzezinska A, et al: Potentiated antitumor effects of the
combination treatment with statins and pamidronate in vitro and in
vivo. Int J Oncol. 30:1413–1425. 2007.PubMed/NCBI
|
|
56
|
Liao J, Chung YT, Yang AL, Zhang M, Li H,
Zhang W, Yan L and Yang GY: Atorvastatin inhibits pancreatic
carcinogenesis and increases survival in
LSL-KrasG12D-LSL-Trp53R172H-Pdx1-Cre mice. Mol Carcinog.
52:739–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fendrich V, Sparn M, Lauth M, Knoop R,
Plassmeier L, Bartsch DK and Waldmann J: Simvastatin delay
progression of pancreatic intraepithelial neoplasia and cancer
formation in a genetically engineered mouse model of pancreatic
cancer. Pancreatology. 13:502–507. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mohammed A, Qian L, Janakiram NB,
Lightfoot S, Steele VE and Rao CV: Atorvastatin delays progression
of pancreatic lesions to carcinoma by regulating PI3/AKT signaling
in p48Cre/+ LSL-KrasG12D/+ mice. Int J Cancer. 131:1951–1962. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mistafa O and Stenius U: Statins inhibit
Akt/PKB signaling via P2X7 receptor in pancreatic cancer cells.
Biochem Pharmacol. 78:1115–1126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ding N, Cui XX, Gao Z, Huang H, Wei X, Du
Z, Lin Y, Shih WJ, Rabson AB, Conney AH, et al: A triple
combination of atorvastatin, celecoxib and tipifarnib strongly
inhibits pancreatic cancer cells and xenograft pancreatic tumors.
Int J Oncol. 44:2139–2145. 2014.PubMed/NCBI
|
|
61
|
Gbelcová H, Svéda M, Laubertová L, Varga
I, Vítek L, Kolář M, Strnad H, Zelenka J, Böhmer D and Ruml T: The
effect of simvastatin on lipid droplets accumulation in human
embryonic kidney cells and pancreatic cancer cells. Lipids Health
Dis. 12:1262013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ishikawa S, Nagai Y, Masuda T, Koga Y,
Nakamura T, Imamura Y, Takamori H, Hirota M, Funakosi A, Fukushima
M and Baba H: The role of oxysterol binding protein-related protein
5 in pancreatic cancer. Cancer Sci. 101:898–905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Souchek JJ, Baine MJ, Lin C, Rachagani S,
Gupta S, Kaur S, Lester K, Zheng D, Chen S, Smith L, et al:
Unbiased analysis of pancreatic cancer radiation resistance reveals
cholesterol biosynthesis as a novel target for radiosensitisation.
Br J Cancer. 111:1139–1149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Maione F, Oliaro-Bosso S, Meda C, Di
Nicolantonio F, Bussolino F, Balliano G, Viola F and Giraudo E: The
cholesterol biosynthesis enzyme oxidosqualene cyclase is a new
target to impair tumour angiogenesis and metastasis dissemination.
Sci Rep. 5:90542015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yang Y, Liu H, Li Z, Zhao Z, Yip-Schneider
M, Fan Q, Schmidt CM, Chiorean EG, Xie J, Cheng L, et al: Role of
fatty acid synthase in gemcitabine and radiation resistance of
pancreatic cancers. Int J Biochem Mol Biol. 2:89–98.
2011.PubMed/NCBI
|
|
66
|
Hussein D and Mo H:
d-δ-Tocotrienol-mediated suppression of the proliferation of human
PANC-1, MIA PaCa-2, and BxPC-3 pancreatic carcinoma cells.
Pancreas. 38:e124–e136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shin-Kang S, Ramsauer VP, Lightner J,
Chakraborty K, Stone W, Campbell S, Reddy SA and Krishnan K:
Tocotrienols inhibit AKT and ERK activation and suppress pancreatic
cancer cell proliferation by suppressing the ErbB2 pathway. Free
Radic Biol Med. 51:1164–1174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Guillaumond F, Bidaut G, Ouaissi M,
Servais S, Gouirand V, Olivares O, Lac S, Borge L, Roques J, Gayet
O, et al: Cholesterol uptake disruption, in association with
chemotherapy, is a promising combined metabolic therapy for
pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 112:2473–2478.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Menter DG, Ramsauer VP, Harirforoosh S,
Chakraborty K, Yang P, Hsi L, Newman RA and Krishnan K:
Differential effects of pravastatin and simvastatin on the growth
of tumor cells from different organ sites. PLoS One. 6:e288132011.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gbelcová H, Lenícek M, Zelenka J, Knejzlík
Z, Dvoráková G, Zadinová M, Poucková P, Kudla M, Balaz P, Ruml T
and Vítek L: Differences in antitumor effects of various statins on
human pancreatic cancer. Int J Cancer. 122:1214–1221. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hong JY, Nam EM, Lee J, Park JO, Lee SC,
Song SY, Choi SH, Heo JS, Park SH, Lim HY, et al: Randomized
double-blinded, placebo-controlled phase II trial of simvastatin
and gemcitabine in advanced pancreatic cancer patients. Cancer
Chemother Pharmacol. 73:125–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Swierczynski J, Hebanowska A and
Sledzinski T: Role of abnormal lipid metabolism in development,
progression, diagnosis and therapy of pancreatic cancer. World J
Gastroenterol. 20:2279–2303. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Søreide K and Sund M:
Epidemiological-molecular evidence of metabolic reprogramming on
proliferation, autophagy and cell signaling in pancreas cancer.
Cancer Lett. 356:281–288. 2015. View Article : Google Scholar : PubMed/NCBI
|














