Introduction
At low levels, nitric oxide (NO•) is a
signaling molecule required for many physiological functions.
However, when produced in excessive amounts, for example, by
inflammatory cells, it can cause cell death and mutagenicity, as
demonstrated by numerous experiments in cultured cells and
experimental animal models (1,2). The
concentration, source and cumulative dosage of NO•, as
well as the composition of biological milieu, can affect cellular
responses. NO• acts on cells and tissues in three ways:
By diffusing into cells and undergoing intracellular consumption,
autoxidating to form nitrous anhydride
(N2O3), and reacting with superoxide
(O2•−) to form peroxynitrite
(ONOO−) (1,2). The reactive oxygen species (ROS) formed
by these reactions may then react further: ONOO− reacts
with carbon dioxide to form nitrosoperoxycarbonate
(ONOOCOO−); N2O3, ONOO−
and ONOOCOO− breakdown to form reactive species such as
hydroxyl radicals (OH•), nitrogen dioxide radicals
(NO2•) and carbonate radical anions
(CO3•−); and all of these species may react
with cellular molecules (1,3–9).
Overproduction of these reactive species under pathological
conditions such as inflammation induces oxidative and nitrosative
stress and damages cellular macromolecules, which increases levels
of mutation and carcinogenesis (3–9).
Adverse responses to NO• reflect
qualitative and quantitative variation among different types of
cells. Apoptosis is the main mechanism by which cell death occurs
at low doses of NO•, whereas necrosis occurs more
readily at higher doses. However, there is a wide variation of
responses to NO• among different cell types. For
example, sub-millimolar concentrations of NO•-donor
drugs rapidly induce cell death in macrophages, however, they have
no obvious effect on cultured hepatocytes. Furthermore, some cell
types, such as human lymphoblastoid cells, are sensitive to
apoptosis, whereas others, including HCT116 human colon cancer
cells, exhibit resistance (2). These
differences may be down to the actions of other factors acting on
the cell. Antioxidants (for example, glutathione and flavonoids)
can alter the apoptosis-inducing potency of different
NO• donors. Other cellular factors, such as enzymatic
scavengers of free radicals (including superoxide dismutase and
catalase) may also contribute to the differences in sensitivity of
cell to NO•-induced cell death.
Factors affecting the mutagenic potency induced by
NO• have not yet been extensively characterized.
Previous studies by our group have demonstrated that NO•
and the ROS derived from it are strongly mutagenic in the
supF shuttle vector pSP189 exposed to large single bolus
doses of ONOO−, NO• gas or unstable donors
(10–13). While providing useful mechanistic
information, exposure under these conditions limits extrapolation
of the data to in vivo pathological states in which these
radicals may play a role. To address this issue, studies aimed at
developing well-defined experimental models that could be exposed
to more pathophysiologically relevant conditions, and defining the
mechanisms responsible for NO• and ROS-associated
genotoxicity have been performed by our group (14–20). The
effects of DNA damage, mutational frequencies and spectra induced
by varying bolus doses of ONOO− were compared with those
induced by slow infusion of ONOO− and by SIN-1, which
slowly decomposes to release NO• and
O2•−, thus producing ONOO−
(14,15). A co-culture system (17,18) was
also employed in which target cells were co-cultivated with mouse
macrophages (RAW 264.7) and stimulated to produce NO• by
interferon-γ (IFN-γ) and lipopolysaccharide (LPS), to characterize
genotoxic responses in endogenous hypoxanthine-guanine
phosphoribosyltransferase (HPRT) and thymidine kinase
(TK1) genes (17) as well as
the transfected supF gene (18). NO•-induced mutagenesis of
the TK1 gene in TK6 (wild-type p53) and NH32 (p53 null)
cells using an NO• reactor specifically designed to
provide tightly controlled steady state concentrations of
NO• and molecular oxygen (O2) have also been
examined (16).
The present study sought to mimic the conditions in
inflamed tissues to assess how NO• and the reactive
species derived from it increase carcinogenic risk. The modulating
influences of additional factors on cytotoxicity and mutagenesis
induced by NO• and associated ROS were assessed using
two delivery methods (reactor and co-culture systems). For
co-culture experiments, a modification of the Costar Transwell™
system was introduced that places target and generator cells into
close proximity (1 µm) whilst facilitating their separation
following treatment. The target cells used for these experiments
were human lymphoblastoid TK6 cells, which have been used
extensively in mutagenicity studies (14,16,21–23).
The relative contribution of NO• to mutagenesis was also
investigated by employing an NO• synthase inhibitor.
Materials and methods
Cell cultures and chemicals
Mouse macrophage-like RAW264.7 cells (American Type
Culture Collection, Manassas, VA, USA) were cultured in high
glucose Dulbecco's modified Eagle's medium (DMEM) containing
L-glutamine supplemented with 100 U/ml penicillin, 100 µg/ml
streptomycin and 10% heat-inactivated fetal bovine serum (Atlanta
Biologicals, Lawrenceville, GA, USA). Cells from the human
lymphoblastoid TK6 cell line were provided by Dr Gerald N. Wogan
(Massachusetts Institute of Technology, Cambridge, MA, USA), and
maintained in RPMI-1640 medium supplemented with antibiotics and
10% heat-inactivated horse serum (Lonza Group Ltd., Basel,
Switzerland). Before each experiment, cell cultures were treated
with CHAT (10 µM 2′-deoxycytidine, 20 µM hypoxanthine, 0.1 µM
aminopterin, and 17.5 µM thymidine) according to a standard
protocol to remove pre-existent mutant cells (21).
Reagents and materials were obtained as follows:
Gases from Air Gas (Edison, NJ, USA); Silastic™ tubing (0.058 in.
i.d., 0.077 in. o.d.) from Dow Corning (Auburn, MI, USA);
NO• synthase and •OH/ONOO−
detection kit from Cell Technology, Inc. (Fremont, CA, USA); total
NO• immunoassay kit and recombinant mouse IFN-γ from
R&D Systems, Inc. (Minneapolis, MN, USA); Escherichia
coli LPS (serotype 0127:B8),
4,5-dihydroxy-1,3-benzene-disulfonic acid (tiron), uric acid,
4-nitroquinoline 1-oxide (4-), 6-thioguanine (6-TG), and
trifluorothymidine (TFT) all from Sigma-Aldrich (St. Louis, MO,
USA); and N-methyl-L-arginine monoacetate (NMA) from
CalBiochem Research (EMD Millipore, Billerica, MA, USA).
Exposure of target cells to
NO• in a reactor system
Cells were exposed to NO• by diffusion
through permeable Silastic™ tubing utilized for specially designed
reactors, with which NO• dose and dose-rate is tightly
controlled at steady state concentrations as described previously
(16,24). TK6 cells, which grow in suspension,
were cultivated with a density of 5×105 cells/ml in 110
ml of culture medium and exposed to NO• at a steady
state concentration of 0.6 µM. The total NO• dose
delivered into the medium was controlled by varying the exposure
time. Cells exposed to argon gas under the same conditions served
as negative controls.
Intracellular NO• and ROS
synthesis in RAW 264.7 cells
Generation of NO• and ROS by RAW 264.7
macrophages was determined using fluorescent probes (Cell
Technology, Inc.). Macrophages were plated in a black, clear bottom
96-well microplate (1×105 cells/well) and incubated for
6 h to allow cells to adhere. Cells were then washed twice with
HBSS and preloaded with dye before incubation at 37°C for 60 min
with either 10 µM diaminofluorescein-2 diacetate (DAF-2DA) for
NO• detection, or 10 µM hydroxyphenyl fluorescein (HPF)
for detection of ROS. After washing twice with HBSS to remove
excess dye, cells were incubated for 8 h in 100 µl of fresh medium
in the presence or absence of 20 U/ml IFN-γ and 20 ng/ml LPS. In
some experiments, NMA, tiron, uric acid or combinations thereof
were also added. After 8 h incubation, fluorescence intensity was
measured in a multi plate Spectra Max Gemini fluorescence reader
(Molecular Devices LLC, Sunnyvale, CA, USA) in triplicate and
averaged every 100 sec for 1 h at 37°C. Excitation and emission
wavelengths were set at 488 and 515 nm, and medium blanks corrected
for auto fluorescence.
Co-culture of TK6 with RAW264.7 cells
in a modified Transwell™ system
Transwell™ permeable supports (Corning Inc., NY,
USA) consist of 100 mm culture dishes each containing a 75 mm
diameter insert carrying a polycarbonate membrane with 0.4 µm pores
that separates two chambers. This design allows the free exchange
of fluids but no direct contact between generator and target cells,
even when both are adherent. Co-culture of TK6 cells with
macrophages was performed by the following modified procedure: i)
The membrane-supporting insert was inverted and macrophages
(1×107 cells) were seeded onto the bottom surface of the
membrane after pretreatment with medium for 1 h at 37°C, ii) After
6 h incubation to allow macrophage attachment, the insert was
restored and the two-chamber unit was assembled in the usual
configuration, iii) 12 ml of culture medium, containing 20 units/ml
IFN-γ and 20 ng/ml LPS, with or without various combinations of 2
mM NMA, 1.25 mM tiron, and 1.25 mM uric acid, was inserted into the
lower chamber of the Transwell™ device, iv) TK6 cells suspended in
9 ml medium were placed into the upper compartment, v) The
concentration of NMA and a nonspecific NOS inhibitor, which were
effective in reducing cytotoxicity (MTT assay, data not shown) was
established using modifications of conditions outlined in Li et
al (14).
Two preliminary experiments were performed to
establish optimal co-culture conditions suitable for assessment of
mutagenic responses. First, it was confirmed that TK6 target doses
do not produce detectable amounts of NO• when cultured
with IFN-γ/LPS for 24 h. Following this, the TK6 cell to macrophage
ratio was established based on NO•-induced cytotoxicity.
In order to evaluate mutagenic responses at equivalent levels of
cell survival, TK6 cells were co-cultured with macrophages at a
ratio of 1:2 (5×106:1×107) in DMEM for 24 h
with shaking to prevent settling of suspended cells. Following
co-culture, TK6 cells were collected, washed twice and re-suspended
in 10 ml of culture medium prior to analysis.
Following each period of co-culture,
NO2− as well as total NO•
(NO3− plus NO2−)
content of cell supernatants were measured using an immunoassay kit
(R&D Systems, Inc.). NO2− was measured by
allowing 50 µl of culture supernatant to react with 100 µl of
Griess reagent at room temperature for 10–30 min. To measure total
NO• production, NADH and NO3−
reductase were added before reaction with the Griess reagent, and
absorbance was measured at 540 nm using a micro plate reader. Total
NO• and NO2− concentrations were
calculated from standard curves derived from standard solutions
provided, with fresh culture media serving as the control.
Cell viability
Cell viability 24 h after treatment was determined
by trypan blue exclusion, which had previously produced results
comparable to those determined by plating efficiency and MTT assay
(18,21).
HPRT mutation assay
Following treatment, TK6 cells were maintained for 7
days to allow phenotypic expression. A total of 24×106
cells were placed into ten 96-well micro titer plates at densities
of 4×104 cells/well in medium containing 2 µg/ml of 6-TG
and TFT to select HPRT and TK1 mutants, respectively.
For plating efficiency, cells from each culture were plated into
96-well dishes at 1 cell/100 µl/well in the absence of selective
agents. After 2 weeks of incubation, colonies were counted and
mutation fractions (MFs) were calculated as described by Li et
al (16). A single HPRT
mutant colony was then transferred to 24 well plates to propagate
mutant cells. Approximately 2×106 mutant cells were
collected for molecular analysis. The spontaneous MF was estimated
from the argon-treated cells for NO• treatment or
untreated cells for co-culture. Cells treated with 4-NQO (140 ng/ml
for 1.5 h) served as positive controls.
Total RNA isolation, RT-PCR for
determining point mutations and intragenic deletions in HPRT
mutants and sequencing
Total RNA from each mutant clone was isolated using
TRI-reagent (Sigma Aldrich) according to the manufacturer's
protocol, and stored at −80°C. 2 µg RNA was used as a template for
the reverse transcription polymerase chain reaction (RT-PCR) with
Omniscript Reverse Transcription kits (Qiagen GimbH, Hilden,
Germany) and the oligo(dT)15 primer (Promega Corporation, Madison,
WI, USA) provided for first-strand cDNA synthesis. The reaction was
performed at 37°C for 1 h to allow the lysis of cell membrane and
the synthesis of first-strand cDNA from polyadenylated mRNA.
Amplification of the cDNA was performed in two rounds of nested PCR
in a PTC-200 DNA Engine Thermal Cycler (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). A 10 µl aliquot of cDNA solution was
transferred into the first round mix of 10 µl 10x PCR buffer, 2 µl
dNTP mix, 0.5 µl taq polymerase, 73.5 µl high powered liquid
chromatography-grade water, 0.2 µl each 25 mM forward (bases −60 to
−41; 5′-CTGCTCCGCCACCGGCTTCC-3′) and reverse (bases −721 to −702;
5′-GATAATTTTACTGGCGATGT-3′) primer and amplified with a PCR profile
of 94°C: 1 min, 30 cycles of 94°C; 1 min, 61°C; 1 min, 72°C; 1 min
and a final extension of 72°C for 7 min. The product from this
reaction was filtered using a Centricon 50 concentrator (Amicon;
EMD Millipore) and resuspended in 100 µl sterile water to avoid
unspecific binding with remaining primers. 10 µl aliquot was used
as template in the second round of PCR using nested primers (bases
−36 to −17; 5′-CCTGAGCAGTCAGCCCGCGC-3′ and bases 701–682;
5′-CAATAGGACTCCAGATGTTT-3′). The PCR conditions were the same in
the second round reaction as those in the first round. The final
product was run on a 1% agarose gel, stained with etidium bromide
and observed under ultraviolet light. For direct sequencing of
HPRT PCR products, the nested PCR products were purified by
a QIAquick PCR Purification Kit (Qiagen GimbH) and aliquots of
these PCR products were then sequenced by the Dana-Farber/Harvard
Cancer Center DNA Resource Core (Boston, MA, USA) using three
primers with the following sequences: Bases 264–252;
5′-ATTTCTATTCAGT-3′, bases 169–181; 5′-ATGGGAGGCCATC-3′ and bases
405–417; 5′-TATAATTGACACT-3′ (Integrated DNA Technologies,
Coralville, IA, USA), using an adapted method (25).
Statistical analysis
All experiments were repeated 2–4 times. The
two-tailed Student's t-test was used for the comparison of test and
control group, and P<0.05 was considered to indicate a
statistically significant difference.
Results
Cytotoxicity and mutagenicity of
NO• in target cells by reactor system
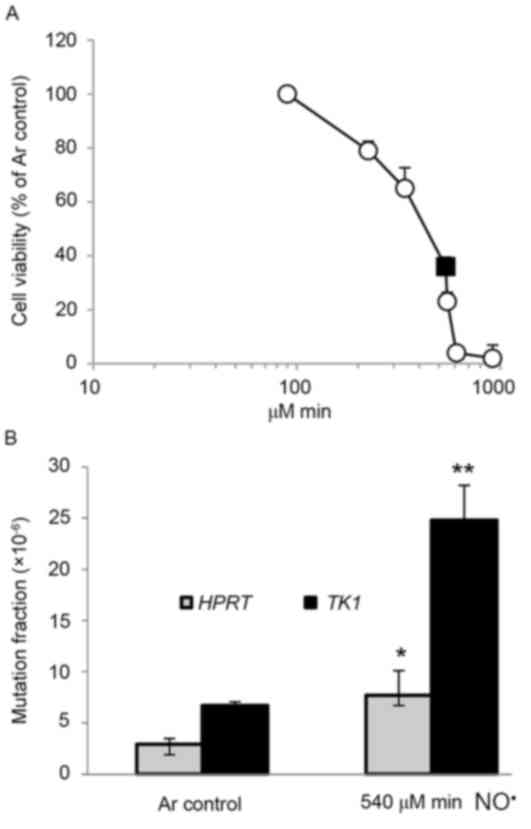
TK6 cells were exposed for 2–24 h to NO•
at a steady state concentration of 0.6 µM, resulting in cumulative
total NO• doses ranging from 90–918 µM/min, which
induced cell death in a dose-dependent manner (Fig. 1A). The cell viability of TK6 cells was
36% 24 h after a dose of 540 µM/min NO• (Fig. 1A). The mutagenicity of NO•
in the HPRT and TK1 genes of TK6 cells was also
investigated (Fig. 1B). At a total
dose of 540 e/min of NO•, induced MFs in the HPRT
and TK1 genes were 7.7×10−6 (P<0.05) and
24.8×10−6 (P<0.01), 2.7- and 3.7-fold higher than
background (2.9×10−6 and 6.7×10−6),
respectively (Fig. 1B). MFs at the
HPRT and TK1 loci in 4-NQO-treated positive controls
were 14.6×10−6 and 28.1×10−6,
respectively.
Intracellular production of
NO• and ROS in activated macrophages
To quantify intracellular NO• and ROS
production by macrophages, DAF-2T and HPF fluorescence intensities
were measured for 1 h, beginning 8 h after activation with
IFN-γ/LPS (Fig. 2). NO•
converts the non-fluorescent dye, DAF-2, to its fluorescent
triazole derivative, DAF-2T (26).
The fluorescence of DAF-2T increased continuously (Fig. 2A), confirming increased NO•
production by macrophages stimulated with IFN-γ/LPS. The probe HPF
was used as an index of the production of ROS, primarily
ONOO− and •OH, which exhibited a modest
increase up to ~30 min, after which production tended to plateau
(Fig. 2B). These experiments indicate
that NO• and ROS were intracellularly generated by
activated macrophages (Fig. 2).
Various treatment regimens were used to provide
additional evidence regarding the reactive species responsible for
these effects, by introducing an inhibitor and scavengers,
including NMA (a NO• synthase inhibitor), tiron (an
O2•− scavenger) and uric acid
(O2•− and ONOO− scavengers), alone
or in combination (NMA+ tiron or NMA+ tiron +uric acid).
Intracellular fluorescence of DAF-2T, i.e., NO•-derived
fluorescence, was almost completely suppressed by NMA, and was
partially suppressed by uric acid (Fig.
2A). By contrast, HPF fluorescence intensity, the ROS probe,
was effectively blocked by uric acid but was not significantly
affected by NMA (Fig. 2B). Results
obtained in the presence of the both probes demonstrated that tiron
and/or uric acid in combination with NMA reduced total fluorescence
almost completely (data not shown), confirming that NO•
and ROS were of central importance as a reactive species produced
by activated macrophages. Additionally, MTT assays performed at the
beginning and end of each experiment confirmed that the observed
changes in fluorescence intensity were not attributable to changes
in cell survival (data not shown).
Viability, NO• production
and mutagenesis in TK6 cells co-cultivated with activated
macrophages
The relative survival, concentration of
NO• and mutation in the HPRT and TK1 genes
of TK6 cells co-cultivated with macrophages for 24 h was determined
(Fig. 3). Activated macrophages
caused reduced (38%) cell survival rates compared to controls
(100%; P<0.01), and 18.3-fold higher total NO•
production compared with untreated controls (P<0.01; Figs. 3A and B). The MFs induced in
HPRT and TK1 genes (8.8×10−6 and
13.9×10−6, respectively) were 2.8- and 1.9-fold higher
than the spontaneous MFs (3.2×10−6 and
7.3×10−6, respectively; P<0.05; Fig. 3C). Co-treatment with both IFN-γ/LPS
and NMA restored cell survival to 95.5%, completely blocked total
NO• and NO2− production and
strongly suppressed the increases in MF by 95 and 89%,
respectively, indicating that NO• was largely
responsible for the cytotoxicity and induced mutations in TK6 cells
(Fig. 3; Table I).
 | Table I.Suppressive effects of NMA on total
NO• and NO2− production,
mutagenesis, and cytotoxicity in TK6 cells co-cultured with
IFN-γ/LPS-stimulated RAW 264.7 cells. |
Table I.
Suppressive effects of NMA on total
NO• and NO2− production,
mutagenesis, and cytotoxicity in TK6 cells co-cultured with
IFN-γ/LPS-stimulated RAW 264.7 cells.
|
| Inhibition (%) |
|
|---|
|
|
|
|
|---|
| Treatment |
Nitrite+Nitrate | Nitrite | Mutation | Cytotoxicity,
% |
|---|
| IFN-γ/LPS | – |
| – | 62.0 |
| IFN-γ/LPS+NMA | 100 | 99.7±1.44 | 94.5±9.32
(HPRT) |
4.5 |
|
|
|
|
|
|
|
|
|
| 89.2±5.69
(TK1) |
|
Molecular analysis of HPRT
mutants
For the molecular analysis of the HPRT
mutations, RT-mediated production of HPRT cDNA, PCR
amplification and cDNA sequencing were used to define small
alterations sin the coding sequence. sRNA was extracted and
analyzed from 22 and 48 mutants in argon and 540 µM min of
NO• treatment groups in the reactor system, and 20 and
51 mutants in untreated and IFN-γ/LPS-treatment groups in the
co-culture system, respectively. Table
II highlights the altered DNA sequences of 15 and 30 cDNA
products synthesized from 22 argon and 48 NO• treated
HPRT mutants, respectively. Single base pair substitutions
(60%) were the major type of argon control mutations, with G:C to
T:A transversions (13%) and G:C to T:A (13%) and A:T to G:C (13%)
transitions. 20% (3/15) of mutants had base deletions and 13%
(2/15) had one complete exon exclusion. Similar mutation profiles
were observed in NO•-induced mutants (Table II). Base substitutions were the
predominant type of mutation (61%), followed by single or two
consecutive exon exclusions (23%) and deletions (13%). Of the 18
mutants carrying single base pair substitutions, an equal number
occurred at G:C (9/18) or A:T (9/18) base pairs with G:C to T:A
(13%) and A:T to T:A (13%) transversions and G:C to A:T (13%) and
A:T to G:C (13%) transitions. Among these, a clustering of base
changes on exons 2 and 4 was observed in comparison to the
spontaneous distribution. Four frame shift mutations, consisting of
1 or 2-bp deletions and 1-bp insertions occurred in exon 3.
| Table II.A summary of results from the
molecular analysis of human HPRT mutant TK6 cells treated
with NO• exposure by reactor or co-culture systems. |
Table II.
A summary of results from the
molecular analysis of human HPRT mutant TK6 cells treated
with NO• exposure by reactor or co-culture systems.
|
| Proportion of
mutants (%) |
|---|
|
|
|
|---|
|
| Reactor | Co-culture |
|---|
|
|
|
|
|---|
|
|
Spontaneousa |
NO•-inducedb |
Spontaneousc |
NO•-inducedd |
|---|
| Transversion | 5
(34) | 10 (35) | 4
(26) | 9
(30) |
| G:C to
T:A | 2
(13) | 4
(13) | 1
(7) | 6
(20) |
| G:C to
C:G | 1
(7) | 1
(3) | 0
(0) | 1
(3) |
| A:T to
T:A | 1
(7) | 4
(13) | 2
(13) | 1
(3) |
| A:T to
C:G | 1
(7) | 1
(3) | 1
(7) | 1
(3) |
| Transition | 4
(26) | 8
(26) | 3
(20) | 7
(24) |
| G:C to
A:T | 2
(13) | 4
(13) | 2
(13) | 2
(7) |
| A:T to
G:C | 2
(13) | 4
(13) | 1
(7) | 5
(17) |
| Insertions | 1
(7) | 1
(3) | 0
(0) | 0
(0) |
| Deletions | 3
(20) | 4
(13) | 1
(7) | 6
(20) |
| Multiple | 0
(0) | 0
(0) | 1
(6) | 1
(3) |
| Exon
exclusions | 2
(13) | 7
(23) | 6
(40) | 7
(23) |
| One
exon | 2
(13) | 1
(3) | 0
(0) | 7
(23) |
| Two
exon | 0
(0) | 6
(20) | 6
(40) | 0
(0) |
| Total | 15 (100) | 30 (100) | 15 (100) | 30 (100) |
In total, 30 of 51 HPRT mutants from TK6
cells co-cultivated with activated macrophages produced HPRT
cDNA. These mutations included 54% (16/30) base substitutions, 23%
(7/30) single exon exclusions and 20% (6/30) deletions (Table II). The proportion of point mutations
at the G:C and A:T base pairs was 56% (9/16) and 44% (7/16),
respectively. The major base substitutions were G:C to T:A
transversions (20%) and A:T to G:C transitions (17%).
NO• derived from co-culture system induced base
substitutions that occurred most frequently in exons 2 and 3 of the
HPRT gene (Table II). 8- to
16-bp deletions at positions between 387 and 402 occurred mostly
frequently in exon 5 and were observed in the co-culture system.
Analysis of spontaneous mutations that formed in the HPRT
gene of TK6 cells co-cultivated with untreated macrophages revealed
that 46% were base substitutions, 7% 1-bp deletions and 40% two
exon exclusions (Table II).
Discussion
It has been previously demonstrated that the
delivery method of NO• and ROS affects cellular
responses, sometimes producing conflicting findings regarding
cytotoxicity, apoptosis and mutation (27,28). The
present study has characterized the mutagenic effects in cells
exposed to NO• and ROS associated with inflammation
through two delivery modes: A reactor system to deliver
NO• by diffusion into cell culture media at tightly
controlled steady state concentrations (16,24); and a
co-culture system to expose target cells to NO• and ROS
generated by activated macrophages. In the first approach,
cytotoxicity and mutagenesis were compared in TK6 cells exposed via
a reactor system for delivering NO• at predictable,
reproducible rates, and induced DNA damage in CHO cells comparable
to those observed in the nuclear DNA of NO•-producing
macrophages (2,29). The results highlighted that
NO• treatment reduced the percentage of viable cells
dose-dependently (Fig. 1A), and
mutagenic responses were characterized at total NO• dose
allowing ~30% cell survival (540 µM/min NO•, closed
square in Fig. 1A). Statistically
significant mutagenic effects of NO• have been observed
previously, however the current study demonstrated that
NO• had a different mutagenic potency at the two
endogenous HPRT and TK1 loci (Fig. 1B), consistent with earlier findings
(16,17,19,21).
Continuous exposure of TK6 cells for 2 h, at a rate of 533 nM/s
NO•, caused a 2-fold increase in HPRT and
4.2-fold in TK1 gene expression as compared with
argon-treated controls (17,21). These differences in susceptibility to
mutations at the two loci may be explained by the fact that
deletions >1.3 Mbp at the HPRT locus decreases cell
viability (30–32), however, cells at the TK1 locus
can tolerate much larger deletions (33). Furthermore, allelic recombination is
possible at the autosomal TK1 locus but not at the X-linked
hemizygous HPRT locus (34).
The present study also characterized the mutagenic
potency of NO• and ROS produced by IFN-γ/LPS activated
macrophages using a modified Transwell™ co-culture system. Before
examining mutagenic events in co-cultivated target cells,
NO• and ROS production were assessed by macrophages
activated with IFN-γ/LPS using fluorescent probes specific for
these factors (Fig. 2). Upon
activation, macrophages produced NO• and ROS, detected
at the intracellular level by specific probes and at the
extracellular level through the formation of
NO3− and NO2−, the
metabolites of NO• in cell supernatants (Fig. 3B; Table
I). These data expand on previous results (35,36),
demonstrating that NO• concentration after 8 h
activation approached a plateau of 0.6 µM at ~34 min as well as a
substantial increase in O2•− production by
activated macrophages. These findings are further substantiated by
decreases in fluorescence intensity of probes specific for
NO• and ROS caused by NMA, an NO• synthase
inhibitor, and O2•− and/or ONOO−
scavengers (Fig. 2).
Previous co-culture studies indicated that
NO• and ROS produced by macrophages were lethal and
strongly mutagenic in both target cells and generator cells
(17,18,20).
Others have demonstrated that products of IFN-γ/LPS-stimulated
RAW264.7 or TPA-stimulated HL-60 cells induced mutagenesis in AS52
cells exposed in mixed co-culture systems (37). As well as increased MF, high levels of
8-hydroxy-2′-deoxyguanosine, reflecting oxidative DNA damage have
been detected in AS52 co-cultivated with both types of generator
cells (37,38). Suppressors of NO• and ROS
strongly inhibited MF (38–41), supporting the conclusion that
NO• and ROS are major causes of induced mutations.
In the present study, a Transwell™ system was used
that permits cell-cell communication through diffusible soluble
factors independent of cell-cell contact, in contrast to the
co-culture systems used in previous studies. These allow direct
generator-target cell contact, potentially maximizing the transfer
of reactive species from generator to target cells, which could
amplify estimates of target cell exposure in inflamed tissues.
Additionally, macrophage and target cells share the same medium
without cell-cell interactions, owing to the physical separation of
cells by a polycarbonate membrane. Using the Transwell™ co-culture
system, activation of macrophages increased production of
NO• and the MF of TK6 cells as compared to unstimulated
controls (Fig. 3; Table I). By employing an NO•
synthase inhibitor, the relative contribution of NO• to
mutagenesis in this system was assessed. Inhibition of
NO• production through addition of NMA to the culture
medium abrogated much of the cytotoxicity and genotoxicity in TK6
cells, confirming the role of NO• in inducing these
effects (Fig. 3; Table I). A contributing factor may have been
distances between NO• generator and target cells, due to
TK6 cells settling out of suspension were less uniformly
distributed than adherent cells growing in close proximity relative
to the monolayer of macrophages. O2•− and
ONOO−-derived ROS have much shorter half-lives (<50
ms) than NO• and shorter diffusion radii, thus they may
not have reached effective concentrations in proximity to TK6 cells
above the monolayer of adherent macrophages (17,18).
Mutagenesis induced by NO• has
previously been examined in a number of experimental systems.
Exposure of S. typhimurium to NO• gas induced G:C
to T:A, T:A to A:T, and C:G to A:T mutations (19), and also mutagenized TK6 cells, causing
A:T to T:A and A:T to G:C mutations in the HPRT gene
(19). Peroxynitrite, the reaction
product of NO• and superoxide, also induced pSP189
supF mutations, predominantly G:C to T:A (10,15). G:C
to T:A mutations occurred in the supF gene of pSP189
replicating in AD293 cells co-cultivated with activated macrophages
(18). Treatment of the same plasmid
with NO• donor drugs (diethylamine/NO• or
spermine/NO• complexes) induced predominantly G:C to A:T
and A:T to G:C mutations (42). In
each of these experimental systems, base substitutions were the
main type of mutations induced, the specific type depended on the
form of NO• used.
In the present study, the majority of spontaneous
and NO• induced mutations were single base pair
substitutions, with transversions (30–35%) outnumbering transitions
(24–26%). Other mutations were deletions and an insertion in both
reactor and co-culture systems (Table
II). 29–30 and 23–29% of mutations occurred at G:C and A:T base
pairs, respectively, and G:C to T:A transversions (13–20%) and A:T
to G:C transitions (13–17%) were the single base pair substitutions
most often induced by NO• treatment in both delivery
systems (Table II). In human cancer,
a significant proportion of mutations in oncogenes and tumor
suppressor genes are G:C to T:A transversions, particularly in lung
cancer (43,44). Previous studies have demonstrated that
these mutations may be caused by a variety of DNA lesions, such as
apurinic sites, 8-oxodeoxyguanosine (8-oxo-dG), or 8-nitroguanine
(8-nitro-G; (45–47). ROS-associated G:C to T:A transversions
may arise from oxidative damage, leading to dA rather than dC being
incorporated opposite dG. A:T to G:C transitions, generally induced
by oxidative damage (19), were also
frequently observed (Table II).
Deamination of adenine to hypoxanthine, which pairs with cytosine
rather than thymine in DNA, could account for the high proportion
of A:T to G:C transitions observed (19). The predominance of G:C to T:A
transversions is consistent with previously reported mutation types
induced by peroxynitrite (ONOO−) and its derivatives
generated by reaction of NO• with
O2•− (10,15,18) and
NO• gas (42), suggesting
that NO• may be a major contributor to the observed
mutagenesis.
To conclude, the present study provides evidence
that exposure to NO• induces cytotoxicity and
mutagenicity in target cells and that inhibition of NO•
production by activated macrophages is effective at abrogating
these properties. Moreover, both the delivery method of
NO• and target genes at which the TK6 cells are exposed
was revealed to strongly influence the cytotoxicity, the mutagenic
potency, types and distribution of mutations. The systems used to
introduce NO• in these experiments were designed to
approximate conditions of exposure physiologically relevant to
chronic inflammation states. Further studies will be required to
elucidate precise mechanisms underlying these effects and their
potential relevance to NO• induced genotoxicity in
vivo.
Acknowledgements
This work was supported by Basic Science Research
Program (no. 2016R1A6A1A03012862 and no. 2014R1A1A2056292) through
the National Research Foundation of Korea (NRF) funded by the
Ministry of Education, Science and Technology, Republic of
Korea.
References
|
1
|
Dedon PC and Tannenbaum SR: Reactive
nitrogen species in the chemical biology of inflammation. Arch
Biochem Biophys. 423:12–22. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li CQ and Wogan GN: Nitric oxide as a
modulator of apoptosis. Cancer Lett. 226:1–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ohshima H, Tatemichi M and Sawa T:
Chemical basis of inflammation-induced carcinogenesis. Arch Biochem
Biophys. 417:3–11. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Czapski G and Goldstein S: The role of the
reactions of NO with superoxide and oxygen in biological systems: A
kinetic approach. Free Radic Biol Med. 19:785–794. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chanock SJ, el Benna J, Smith RM and
Babior BM: The respiratory burst oxidase. J Biol Chem.
269:24519–24522. 1994.PubMed/NCBI
|
|
6
|
Ohshima H and Bartsch H: Chronic
infections and inflammatory processes as cancer risk factors:
Possible role of nitric oxide in carcinogenesis. Mutat Res.
305:253–264. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vamvakas S and Schmidt HH: Just say NO to
cancer? J Natl Cancer Inst. 89:406–407. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wiseman H and Halliwell B: Damage to DNA
by reactive oxygen and nitrogen species: Role in inflammatory
disease and progression to cancer. Biochem J. 313:17–29. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
MacMicking J, Xie QW and Nathan C: Nitric
oxide and macrophage function. Annu Rev Immunol. 15:323–350. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Juedes MJ and Wogan GN:
Peroxynitrite-induced mutation spectra of pSP189 following
replication in bacteria and in human cells. Mutat Res. 349:51–61.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jeong JK, Juedes MJ and Wogan GN:
Mutations induced in the supF gene of pSP189 by hydroxyl radical
and singlet oxygen: Relevance to peroxynitrite mutagenesis. Chem
Res Toxicol. 11:550–556. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tretyakova NY, Burney S, Pamir B, Wishnok
JS, Dedon PC, Wogan GN and Tannenbaum SR: Peroxynitrite-induced DNA
damage in the supF gene: Correlation with the mutational spectrum.
Mutat Res. 447:287–303. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pamir B and Wogan GN: Carbon dioxide
modulation of peroxynitrite-induced mutagenesis of the supF gene in
pSP189. Chem Res Toxicol. 16:487–492. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li CQ, Trudel LJ and Wogan GN:
Genotoxicity, mitochondrial damage and apoptosis in human
lymphoblastoid cells exposed to peroxynitrite generated from SIN-1.
Chem Res Toxicol. 15:527–535. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim MY, Dong M, Dedon PC and Wogan GN:
Effects of peroxynitrite dose and dose rate on DNA damage and
mutation in the supF shuttle vector. Chem Res Toxicol. 18:76–86.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li CQ, Pang B, Kiziltepe T, Trudel LJ,
Engelward BP, Dedon PC and Wogan GN: Threshold effects of nitric
oxide-induced toxicity and cellular responses in wild-type and
p53-null human lymphoblastoid cells. Chem Res Toxicol. 19:399–406.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhuang JC, Lin D, Lin C, Jethwaney D and
Wogan GN: Genotoxicity associated with NO production in macrophages
and co-cultured target cells. Free Radic Biol Med. 33:94–102. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim MY and Wogan GN: Mutagenesis of the
supF gene of pSP189 replicating in AD293 cells cocultivated with
activated macrophages: Roles of nitric oxide and reactive oxygen
species. Chem Res Toxicol. 19:1483–1491. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhuang JC, Wright TL, deRojas-Walker T,
Tannenbaum SR and Wogan GN: Nitric oxide-induced mutations in the
HPRT gene of human lymphoblastoid TK6 cells and in Salmonella
typhimurium. Environ Mol Mutagen. 35:39–47. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhuang JC, Lin C, Lin D and Wogan GN:
Mutagenesis associated with nitric oxide production in macrophages.
Proc Natl Acad Sci USA. 95:8286–8291. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li CQ, Trudel LJ and Wogan GN: Nitric
oxide-induced genotoxicity, mitochondrial damage and apoptosis in
human lymphoblastoid cells expressing wild-type and mutant p53.
Proc Natl Acad Sci USA. 99:10364–10369. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li CQ, Robles AI, Hanigan CL, Hofseth LJ,
Trudel LJ, Harris CC and Wogan GN: Apoptotic signaling pathways
induced by nitric oxide in human lymphoblastoid cells expressing
wild-type or mutant p53. Cancer Res. 64:3022–3029. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li CQ, Wright TL, Dong M, Dommels YE,
Trudel LJ, Dedon PC, Tannenbaum SR and Wogan GN: Biological role of
glutathione in nitric oxide-induced toxicity in cell culture and
animal models. Free Radic Biol Med. 39:1489–1498. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang C and Deen WM: Nitric oxide delivery
system for cell culture studies. Ann Biomed Eng. 31:65–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang JL, Maher VM and McCormick JJ:
Amplification and direct nucleotide sequencing of cDNA from the
lysate of low numbers of diploid human cells. Gene. 83:347–354.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hsie AW, Recio L, Katz DS, Lee CQ, Wagner
M and Schenley RL: Evidence for reactive oxygen species inducing
mutations in mammalian cells. Proc Natl Acad Sci USA. 83:9616–9620.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Patel RP, McAndrew J, Sellak H, White CR,
Jo H, Freeman BA and Darley-Usmar VM: Biological aspects of
reactive nitrogen species. Biochim Biophys Acta. 1411:385–400.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miller MR and Megson IL: Recent
developments in nitric oxide donor drugs. Br J Pharmacol.
151:305–321. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tamir S, Lewis RS, de Rojas Walker T, Deen
WM, Wishnok JS and Tannenbaum SR: The influence of delivery rate on
the chemistry and biological effects of nitric oxide. Chem Res
Toxicol. 6:895–899. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nelson SL, Jones IM, Fuscoe JC,
Burkhart-Schultz K and Grosovsky AJ: Mapping the end points of
large deletions affecting the hprt locus in human peripheral blood
cells and cell lines. Radiat Res. 141:2–10. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Phillips EN, Xia F, Kelsey KT and Liber
HL: Spectra of spontaneous and X-ray-induced mutations at the hprt
locus in related human lymphoblast cell lines that express
wild-type or mutant p53. Radiat Res. 143:255–262. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamada Y, Park MS, Okinaka RT and Chen DJ:
Molecular analysis and comparison of radiation-induced large
deletions of the HPRT locus in primary human skin fibroblasts.
Radiat Res. 145:481–490. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Giver CR and Grosovsky AJ: Radiation
specific patterns of loss of heterozygosity on chromosome 17q.
Mutat Res. 450:201–209. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wiese C, Gauny SS, Liu WC,
Cherbonnel-Lasserre CL and Kronenberg A: Different mechanisms of
radiation-induced loss of heterozygosity in two human lymphoid cell
lines from a single donor. Cancer Res. 61:1129–1137.
2001.PubMed/NCBI
|
|
35
|
Lewis RS, Tamir S, Tannenbaum SR and Deen
WM: Kinetic analysis of the fate of nitric oxide synthesized by
macrophages in vitro. J Biol Chem. 270:29350–29355. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nalwaya N and Deen WM: Nitric oxide,
oxygen, and superoxide formation and consumption in macrophage
cultures. Chem Res Toxicol. 18:486–493. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim HW, Murakami A, Williams MV and
Ohigashi H: Mutagenicity of reactive oxygen and nitrogen species as
detected by co-culture of activated inflammatory leukocytes and
AS52 cells. Carcinogenesis. 24:235–241. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Murakami A, Takahashi D, Kinoshita T,
Koshimizu K, Kim HW, Yoshihiro A, Nakamura Y, Jiwajinda S, Terao J
and Ohigashi H: Zerumbone, a Southeast Asian ginger sesquiterpene,
markedly suppresses free radical generation, proinflammatory
protein production and cancer cell proliferation accompanied by
apoptosis: The alpha,beta-unsaturated carbonyl group is a
prerequisite. Carcinogenesis. 23:795–802. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim HW, Murakami A, Abe M, Ozawa Y,
Morimitsu Y, Williams MV and Ohigashi H: Suppressive effects of
mioga ginger and ginger constituents on reactive oxygen and
nitrogen species generation and the expression of inducible
pro-inflammatory genes in macrophages. Antioxid Redox Signal.
7:1621–1629. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim HW, Murakami A, Williams MV and
Ohigashi H: Suppressive effects of selected antioxidants on the
activated leukocytes-induced mutagenesis in the co-culture assay
systems. Biosci Biotechnol Biochem. 68:238–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim HW, Murakami A, Nakamura Y and
Ohigashi H: Screening of edible Japanese plants for suppressive
effects on phorbol ester-induced superoxide generation in
differentiated HL-60 cells and AS52 cells. Cancer Lett. 176:7–16.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Routledge MN, Wink DA, Keefer LK and
Dipple A: DNA sequence changes induced by two nitric oxide donor
drugs in the supF assay. Chem Res Toxicol. 7:628–632. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bennett WP, Hussain SP, Vahakangas KH,
Khan MA, Shields PG and Harris CC: Molecular epidemiology of human
cancer risk: Gene-environment interactions and p53 mutation
spectrum in human lung cancer. J Pathol. 187:8–18. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hainaut P and Pfeifer GP: Patterns of p53
G→T transversions in lung cancers reflect the primary mutagenic
signature of DNA-damage by tobacco smoke. Carcinogenesis.
22:367–374. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang D, Kreutzer DA and Essigmann JM:
Mutagenicity and repair of oxidative DNA damage: Insights from
studies using defined lesions. Mutat Res. 400:99–115. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shibutani S, Takeshita M and Grollman AP:
Insertion of specific bases during DNA synthesis past the
oxidation-damaged base 8-oxodG. Nature. 349:431–434. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Moriya M: Single-stranded shuttle phagemid
for mutagenesis studies in mammalian cells: 8-oxoguanine in DNA
induces targeted G.C->T.A transversions in simian kidney cells.
Proc Natl Acad Sci USA. 90:1122–1126. 1993. View Article : Google Scholar : PubMed/NCBI
|