Introduction
Human aspartyl-(asparaginyl) β-hydroxylase (HAAH) is
a highly conserved enzyme belonging to the
α-ketoglutarate-dependent dioxygenase family. HAAH catalyzes the
β-hydroxylation of aspartyl and asparaginyl residues in epidermal
growth factor (EGF)-like repeats of certain proteins including
Notch, Jagged and Delta-like (1,2). HAAH is
highly specific for malignant neoplasms, including hepatocellular
carcinoma, lung, pancreatic, colorectal and neural carcinomas
(3–5);
however, HAAH exhibits minimal expression in normal tissues
(6,7).
HAAH can be detected in the sera of patients with breast, colon,
lung and prostate cancers, and has been developed into early
diagnostic kits, such as the Panacea HAAH blood tests (BC
Detect® CC Detect®, LC
Detect® and PC Detect®; Panacea
Global, Inc., Richmond Hill, ON, Canada; http://www.panaceaglobalinc.com/panacea-haah-blood-test.html).
HAAH may have a potential role in inducing cellular
transformation and increasing cell motility and invasiveness, which
is required for tumor cell infiltration and metastasis (3,6). The
potential in the application of this tumor-associated antigen as a
biomarker for tumor diagnosis and treatment has been the subject of
several recent investigations (8–10).
HAAH is a type 2 transmembrane protein which can be
divided into four distinct domains: A cytoplasmic amino-terminal
domain, a transmembrane domain, a negatively charged domain that
projects into the lumen of the endoplasmic reticulum and a
catalytic carboxyl terminal domain containing dibasic glycine and
His2 motifs; these have all been previously demonstrated to be
critical for the aspartyl hydroxylase catalytic activity (2,7). In our
previous study, the N-terminal domain of HAAH (HAAH-N), which is
responsible for its biological activity, was successfully expressed
in Escherichia coli (E. coli), and a monoclonal antibody
(mAb) against HAAH-N was obtained (11). Subsequently, this N-terminal specific
mAb was applied in the detection of HAAH overexpression and
distribution in tumor tissues and cells (8), and exhibited positive specificity and
sensitivity to HAAH and its isoform. However, as a potential target
and biomarker for tumor diagnosis, the molecular function of HAAH
in tumor invasion and metastasis requires further study to be fully
understood, and to develop more potent and efficient immunological
tools. The present study primarily described a method for the
expression and purification of the C-terminal domain of HAAH, using
the Pichia pastoris expression system in a 10-L bioreactor.
In addition, this recombinant protein was used as an immunogen to
prepare an mAb against the HAAH C-terminal (HAAH-C).
Immunofluorescence was used to demonstrate the specificity of this
novel antibody. The antibody-dependent cellular cytotoxicity (ADCC)
of natural killer (NK) cells on this antibody was also assessed.
Finally, the novel HAAH-C antibody was used to establish a double
antibody sandwich enzyme-linked immunosorbent assay (ELISA) method
with the previously obtained HAAH-N antibody, and to analysis the
HAAH content in the culture supernatant of carcinoma cell
lines.
Materials and methods
Expression and purification of
recombinant HAAH-C (rHAAC-C)
HAAH cDNA was obtained using an oligo dT primer
(GenScript, Nanjing, China) as described in a previous study
(8,11,12). A
Pichia expression kit containing the P. pastoris strain
GS115 (American Type Culture Collection, Manassas, VA, USA)
and the Invitrogen pPIC9k vector (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) were used to clone the HAAH-C gene.
Oligonucleotide primers, including HAAH-C-F, which contained an
EcoRI restriction enzyme site (5′-CTGAATTCATGAGAGGTTCCCTGCAGA-3′)
and HAAH-C-R, which contained a NotI restriction enzyme site
(5′-TAGCGGCCGCTTAAATTGCTGGAAGGCTGCG-3′),
were designed using prior published sequences from GenBank
(GI:14589865) and used for the amplification of a truncated HAAH
gene (969 bp), which encoded a 38 kDa truncated protein. The
rHAAH-C was expressed in the P. pastoris expression system
and induced with methanol in a 10-L Biostat B plus bioreactor
(Sartorius AG, Göttingen, Germany). The rHAAH-C in the culture
supernatant was purified using the Labscale TFF System (EMD
Millipore, Billerica, MA, USA), Sephadex G25 gel-filtration column
and DEAE Sepharose FF column (GE Healthcare Bio-Sciences,
Pittsburgh, PA, USA), following the manufacturer's
instructions.
For SDS-PAGE analysis, the proteins in the culture
supernatants were mixed with 2X loading buffer (pH 6.8) containing
1 M Tris, 20% glycerol, 10% SDS, 0.1% bromophenol blue and 5%
β-mercaptoethanol. A low molecular weight range ladder (Takara Bio,
Inc., Otsu, Japan) was used as a standard to evaluate the protein
molecular masses. Electrophoresis was carried out on a 12%
polyacrylamide gel under denaturing conditions for ~90 min with a
constant voltage of 120 V. The protein bands were visualized with
Coomassie brilliant blue R-250 staining.
For the western blot analysis, the fractionated
proteins were transferred onto nitrocellulose membranes (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) by electroblotting and
probed with a diluted (1:1,000) anti-HAAH polyclonal antibody
(#CSB-PA002226GA01HU; CUSABIO, Wuhan, China) at 37°C for 1 h. This
was followed by incubation with a goat anti-rabbit immunoglobulin
(Ig)G/horseradish peroxidase (HRP; dilution, 1:2,000; Caltag
Laboratories, Caltag Medsystems, Buckingham, UK) as the secondary
antibody. The western blots were blocked, washed, and probed at
room temperature in 10 mM sodium phosphate (pH 7.4), containing 150
mM NaCl, 0.1% bovine serum albumin (BSA) (Gibco; Thermo Fisher
Scientific, Inc.) and 0.1% Tween 20. The detection of rHAAH-C was
performed using the Enhanced Chemiluminescence Western Blotting
Substrate kit (Pierce; Thermo Fisher Scientific, Inc.).
Generation, purification and
characterization of a mAb against rHAAH-C
The desalted and lyophilized rHAAH-C protein was
purified using a DEAE Sepharose FF column and weighted and diluted
with phosphate buffered saline (PBS) to a concentration of 1 mg/ml;
this was subsequently used as an immunogen. For the initial
immunization, five female BALB/c mice (age, 6–7 weeks; weight,
22–25 g) were obtained from the Laboratory Animal Center of The
Fourth Military Medical University (Xi'an, China) and housed in a
specific pathogen-free environment. The mice were subcutaneously
vaccinated with 100 µg of the immunogen, which was emulsified with
an equal volume of complete Freund's adjuvant (Sigma-Aldrich, St.
Louis, MO, USA). Subsequent booster injections were administered
intraperitoneally, with the same quantity of immunogen, at two and
four weeks post initial injection. The antiserum of each mouse was
collected from the retrobulbar plexus and indirect ELISA determined
each antiserum titer. The best-performing mouse was selected for
hybridoma production and boosted with 100 µg of the immunogen two
days prior to cell fusion.
Mouse myeloma Sp2/0 cells were prepared to a
concentration of 4×105 cells/ml (exponential growth
phase) prior to cell fusion. The harvested spleen cells from the
immunized mice were combined with Sp2/0 cells at a ratio of 10:1
and centrifuged at 300 × g for 8 min at room temperature.
The pellet was then washed twice and centrifuged again at 300 ×
g for 8 min at room temperature. The chamber of a
Micro-Pulser Electroporator (Bio-Rad Laboratories, Inc.) was filled
with the mixed cells and fusion was conducted immediately. The
electro-fusion mode was as follows: Pre-alignment voltage, 5 V
(duration, 30 sec); pulse voltage, 20–30 V (duration, 15 msec); and
post-alignment voltage, 5 V (duration, 30 sec). Following fusion,
the chambers were allowed to stand for 10 min at room temperature.
The chamber was unscrewed and the electrode core rinsed with 1 ml
of post-fusion medium (RPMI-1640 culture medium supplemented with
10% fetal calf serum, 10 mM nonessential amino acids, 100 IU/ml of
penicillin and 100 µg/ml of streptomycin) in the electrode beaker.
The hybridomas were selectively cultured for approximately two
weeks, and an indirect ELISA screened the resulting culture
supernatants. The hybridomas that produced antibodies with a good
reactivity against HAAH-C were subsequently cloned twice more by
limiting dilution, followed by expansion for the large-scale
production of the mAb.
Following the injection of the hybridoma cells
(5×105), ascites was observed in BALB/c mice from 7–14
days. The fluids were purified using a Protein G Sepharose 4 Fast
Flow column (GE Healthcare Bio-Sciences), and the purity was
analyzed by SDS-PAGE, as aforementioned. Isotyping of the mAbs
against HAAH-C was determined using a gel gold test strip mouse mAb
isotyping kit (Pierce; Thermo Fisher Scientific, Inc.) following
the manufacturer's recommendations.
Cells
The cell lines of human cervical cancer (HeLa),
breast carcinoma (MCF-7), liver hepatocellular carcinoma (HepG2)
and mouse myeloma cells lines (Sp2/0) were purchased from the
American Type Culture Collection. The cells were maintained in
Dulbecco's modified Eagle's medium (DMEM) or RPMI-1640 culture
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% fetal calf serum (Gibco; Thermo Fisher Scientific, Inc.) that
had been heat-inactivated at 56°C for 30 min, 10 mM nonessential
amino acids, 100 IU/ml of penicillin and 100 µg/ml of streptomycin
(Genview, Carlsbad, CA, USA), in a humidified 5% CO2
atmosphere at 37°C.
Human NK cells were expanded and cultured as
described previously (13). Briefly,
peripheral venous blood (10 ml) from healthy donors (n=10; 3
females and 7 males; date, 14th March, 2014; Hospital of
Northwestern Polytechnical University, Xi'an, China) was collected
in heparinized tubes. The procedure of the blood sample collection
conformed to the informed consent guidelines of the Ethics
Committee of Northwestern Polytechnical University. The peripheral
blood mononuclear cells (PBMCs) were collected using Lymphocyte
Separation Liquid (Haoyang TBD, Tianjin, China). Following two
washes with PBS the PBMCs were resuspended in RPMI-1640 media,
which was supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) containing 100 IU of interleukin-2
(Peprotech, Inc., Rocky Hill, NJ, USA), 100 µg/ml of penicillin and
100 µg/ml of streptomycin (Genview, Carlsbad, CA, USA). The PBMCs
were counted and cocultured with stimulating cells (13). The CD56-PE and CD3-FITC mAbs, and
their isotype-matched controls (IgG1-FITC/IgG2-PE; QuantoBio
Biotechnology Co., Ltd., Beijing, China), were used to test the
percentage of NK cells (CD56+CD3−) in the
PBMC suspensions after three weeks of ex vivo-expansion,
using flow cytometry (BD FACSCalibur; BD Biosciences, San Jose, CA,
USA).
Immunofluorescence cell staining
Hybridoma cells (4×105) in 3 ml culture
medium were seeded into 6-well cell culture plates and incubated
overnight at 37°C. After washing three times with ice-cold PBS, the
cells were permeabilized for 30 min with 2% Triton X-100 at room
temperature and then blocked with 3% BSA at 37°C for 30 min. The
mAb against HAAH-C (dilution, 1:100; 100 µg/ml) was applied for 1 h
at 37°C, followed by a PBS wash for 3 min. The cells were incubated
with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse
IgG (#sc-2010; Santa Cruz Biotechnology, Inc., Dallas, Texas, USA;
dilution, 1:200) and propidium iodide (PI) at 37°C for 1 h in the
dark. After washing with PBS, the cells were placed with a 50%
glycerol/PBS mounting medium. Images were immediately observed and
captured using fluorescent microscopy.
ADCC assay
The ADCC activity of human NK cells on the HAAH-C
mAb was measured by calculating the rates at which NK cells killed
target cells in a 96-well plate. NK cells were expanded and
cultured as previously described (10,13). The
target cells (HeLa, MCF-7 and HepG2) were incubated with NK cells
as an effector, to a target ratio of 10:1 in the presence or
absence of the anti-HAAH-C mAb (1 µg/ml). NK cells (100 µl;
2×105 cells) were plated into each well of a 96-well
plate and mixed with 100 µl of the target cells (2×104).
Each experiment was performed in triplicate. The NK cell effector
control wells contained 100 µl NK cells (2×105) and 100
µl RPMI-1640 medium. The target cell control wells contained 100 µl
target cells (2×104) and 100 µl RPMI-1640 medium. The
plate was incubated at 37°C for 4 h in a 5% CO2
atmosphere; subsequently, 20 µl Cell Counting Kit-8 (Dojindo
Molecular Technologies, Inc., Shanghai, China) was added to each
well and the plate was then incubated for another 2 h. The
absorbance (A) values were recorded at 450 nm, and the killing rate
of NK cells compared with the target cells was calculated with the
following equation (14):
Killingrate(%)=[1–(Ae+t–Ae)/At]x100%
In this equation, Ae is the mean
A450 of triplicate wells for the NK cell control;
At, is the mean A450 of
triplicate wells for the target cell control; and
Ae+t is the mean A450 of
triplicate wells for NK cells plus target cells.
Double antibody sandwich-ELISA
The anti-HAAH-C mAb was used as capture antibody,
whereas anti-HAAH-N mAb was used as detection antibody in the
double antibody sandwich-ELISA. The culture supernatant of three
tumor cells lines (HeLa, MCF-7, HepG2) and NK cells (negative
control) were used as test samples. The dilution of the capture
antibody, detection antibody, test samples and goat anti-mouse
IgG-HRP conjugate (#ab97023; Abcam, Shanghai, China) were optimized
by checkerboard titration. Sandwich ELISA was performed following
the method described in literature (15). Anti-HAAH-C mAb was diluted in a
coating buffer (0.05 M carbonate-bicarbonate buffer, pH 9.6) and
coated microtiter plates at 4°C overnight. After washing three
times with PBS containing 0.05% (v/v) Tween-20 (PBST), the wells
were blocked with 5% BSA (pH 7.2) at room temperature for 2 h.
Subsequently, the plates were incubated at 37°C for 1 h with 100 µl
of diluted samples. After washing six times with PBST, anti-HAAH-N
mAb was added into each well to detect the antigens (HAAH in
culture supernatant). The wells were washed again with PBST to
remove unbound antibodies, and then the plates were incubated with
HRP-conjugated goat anti-mouse IgG antibody diluted in diluent
buffer at 37°C for 1 h. The bound-antibodies were detected by
dispensing a TMB HRP Color Development Solution (Beyotime Institute
of Biotechnology, Shanghai, China). Finally the absorbance was
measured at 450 nm in microplate reader (BioTek Synergy-4). rHAAH
protein (Cusabio, Wuhan, China) at various concentrations (400,
200, 100, 50, 25, 12.50, 6.25, 3.13, 1.56, and 0 ng/ml) in diluent
buffer was used to generate a standard curve for evaluating test
samples quantitatively. The cut-off value was defined as the mean
value plus three standard deviations (SD) of the mean A value
obtained from RPMI-1640 culture medium (negative control).
Statistical analysis
Statistical analysis was performed using SPSS 16.0
statistical software (SPSS, Inc., Chicago, IL USA). The data are
presented as the mean ± standard deviation. The results were
analyzed using the analysis of variance. Multiple comparisons used
a least significant difference test to evaluate the significance of
differences between groups. P<0.05 was considered to indicate a
statistically significant result.
Results
Expression and purification of
rHAAH-C
The high-level expression of HAAH-C by fermentation
was terminated at 118 h, with a cell wet-weight of 521 g/l in the
10-L bioreactor. It was revealed that the highest expression of
38-kDa rHAAH-C was at 96 h post methanol induction (Fig. 1A). Analysis of the western blot
indicated that the rHAAH-C protein reacted markedly with the
polyclonal antibody against HAAH and exhibited a high specificity
(Fig. 1B).
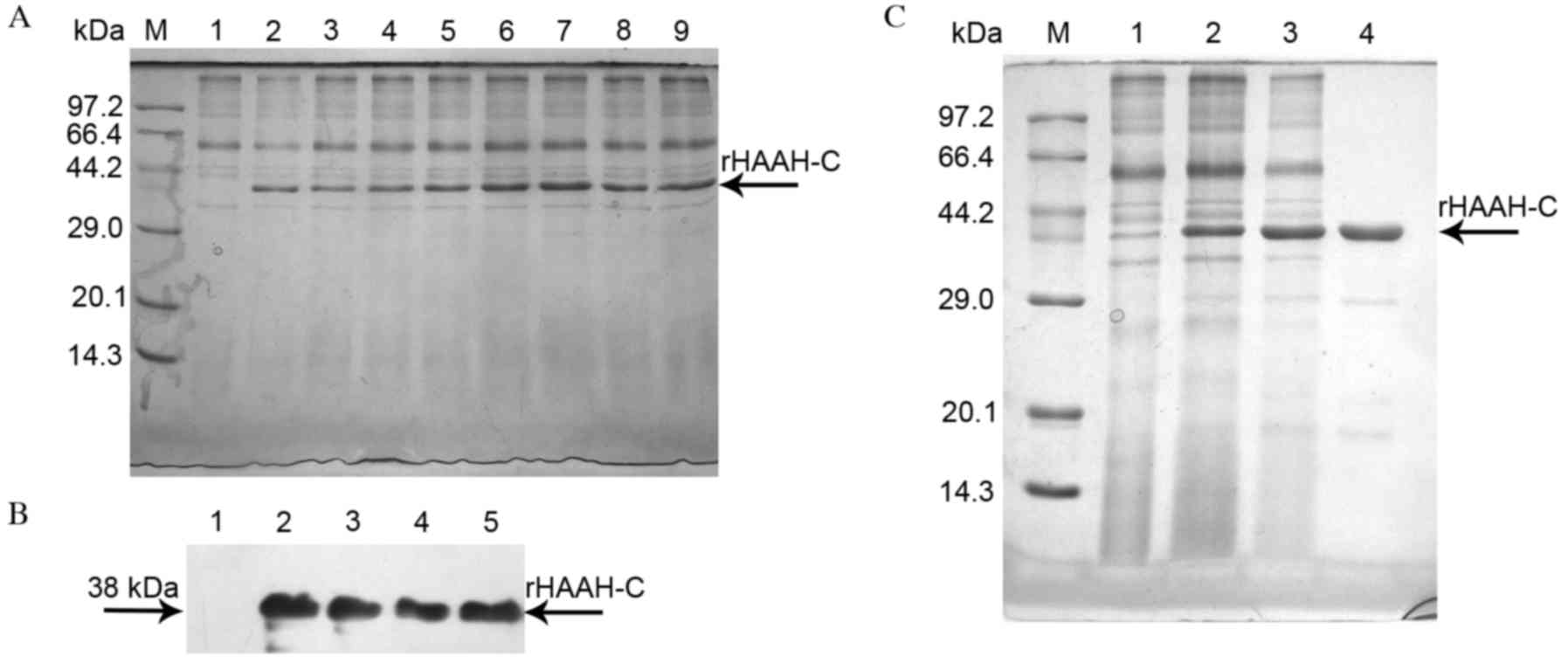 | Figure 1.Characterization and purification of
rHAAH-C protein. (A) SDS-PAGE analysis of the expression of rHAAH-C
in high cell-density fermentation. Lane M, a molecular weight
standard; lanes 1–9, rHAAH-C (fermentation times: lane 1, 0 h; lane
2, 12 h; lane 3, 24 h; lane 4, 36 h; lane 5, 48 h; lane 6, 72 h;
lane 7, 96 h; lane 8, 108 h; lane 9, 120 h). The main recombinant
protein band is denoted with an arrow. (B) Western blot analysis of
the expression of rHAAH-C in high cell-density fermentation. Lane
1, a sample of the whole culture supernatant prior to methanol
induction (negative control); lanes 2–5, parallel samples of the
whole culture supernatant following methanol induction. The samples
were probed with an anti-HAAH polyclonal antibody. (C) SDS-PAGE
analysis of the purification of rHAAH-C expressed in P.
pastoris. Lane M, a molecular weight standard; lane 1, negative
control for the secreted protein; lane 2, crude secreted protein;
lane 3, protein purified using Sephadex G25 gel-filtration
chromatography; lane 4, protein purified using DEAE ion-exchange
chromatography. HAAH, human aspartyl-(asparaginyl)-β-hydroxylase;
SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel
electrophoresis; rHAAC-C, recombinant HAAH C-terminal; P.
pastoris, Pichia pastoris. |
All the purification steps were monitored through
SDS-PAGE. The cell-free supernatant was concentrated by
ultrafiltration and purified on a Sephadex G25 gel-filtration
column. Fractions of certain protein impurities were pooled,
concentrated and purified using a DEAE Sepharose FF column
(Fig. 1C). The purification procedure
resulted in the recovery of 92.4 mg of rHAAH-C from the culture
supernatant, accounting for ~53.8% of the total protein content
(Table I).
 | Table I.Purification of recombinant human
aspartyl-(asparaginyl) β-hydroxylase C-terminal. |
Table I.
Purification of recombinant human
aspartyl-(asparaginyl) β-hydroxylase C-terminal.
| Fraction | Protein (mg/l) | Yield (%) |
|---|
| Cell-free
supernatant | 225.8 |
100 |
|
Ultrafiltration | 181.9 | 80.6 |
| DEAE Sepharose FF
column | 127.1 | 56.2 |
| Sephadex G25
gel-filtration column |
92.4 | 40.9 |
Generation, purification and
characterization of an mAb against rHAAH-C
Following the initial injection and three subsequent
boosters, the maximum titer of the anti-HAAH-C mAb purified from
the sera of the immunized mice was approximately 5×103,
as determined by an indirect ELISA (data not shown). The highest
reacting mouse was given a fourth booster and selected for
hybridoma fusion, which increased the titer to 1×104
(data not shown). The fusion cells were seeded in a 96-well culture
plate. Following a two-week culture, the hybridoma cell clones
formed in 85 of the plate wells, yielding a fusion ratio of 88.5%.
Seven of the hybridoma clones in the aforementioned 85 wells were
selected on the basis of their notable ELISA reactivities with the
HAAH-C protein and subsequently subjected to cloning procedures.
Four of these clones (A3, A6, C9, and E4), which exhibited the best
titers, affinities, and cell growth, were finally selected for
limiting dilution.
Purification of the ascites fluid was performed
using a Protein G Sepharose 4 Fast Flow column to 95% homogeneity,
as assessed by SDS-PAGE (Fig. 2A). An
indirect ELISA of the ascites fluid indicated that the titers of
the mAbs specific against rHAAH-C ranged from 5×103 to
1.5×104. These mAbs were able to specifically react with
rHAAH-C as determined by the western blot analysis (Fig. 2B). A mouse mAb isotyping test kit
determined that the Ig subclasses of the mAbs secreted by the four
cell strains were all IgG1κ. The affinity constants of the four
mAbs ranged from 1.7×108 to 1.2×109, as
determined by a noncompetitive ELISA (Table II).
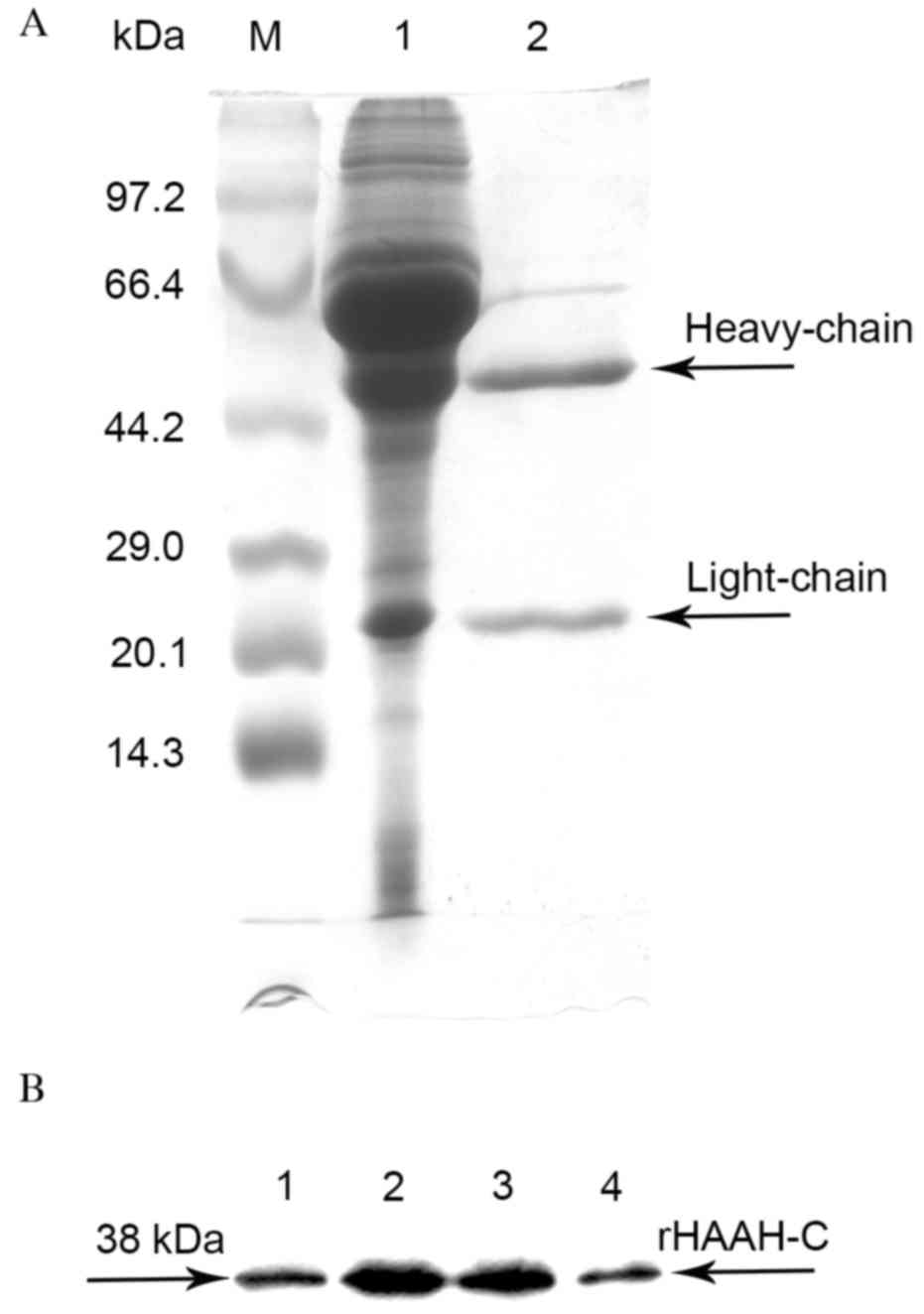 | Figure 2.SDS-PAGE and western blot analysis of
the purification of the mAbs against rHAAH-C. (A) A time course of
the whole culture supernatant. Lane M, a molecular weight standard;
lane 1, the ascites fluid; lane 2, the eluate sample of the mAbs
against rHAAH-C (heavy chain and light chain); (B) Western blot
analysis of the rHAAH-C mAbs. Lane 1, a sample probed with the mAb
A3; lane 2, a sample probed with the mAb A6; lane 3, a sample
probed with the mAb C9; lane 4, a sample probed with the mAb E4.
HAAH, human aspartyl-(asparaginyl)-β-hydroxylase; rHAAC-C,
recombinant HAAH C-terminal; mAbs, monoclonal antibodies. |
| Table II.Identification and characterization
of anti-human aspartyl-(asparaginyl) β-hydroxylase C-terminal
monoclonal antibodies. |
Table II.
Identification and characterization
of anti-human aspartyl-(asparaginyl) β-hydroxylase C-terminal
monoclonal antibodies.
| Hybridoma | Class and
subclass | Type | Titer of
supernatant of ascites | Affinity constant
(M−1) |
|---|
| A3 |
IgG1 | κ | 1:5,000 |
1.7×108 |
| A6 |
IgG1 | κ | 1:10,000 |
4.6×108 |
| C9 |
IgG1 | κ | 1:15,000 |
1.2×109 |
| E4 |
IgG1 | κ | 1:10,000 |
2.2×108 |
Immunofluorescence staining in
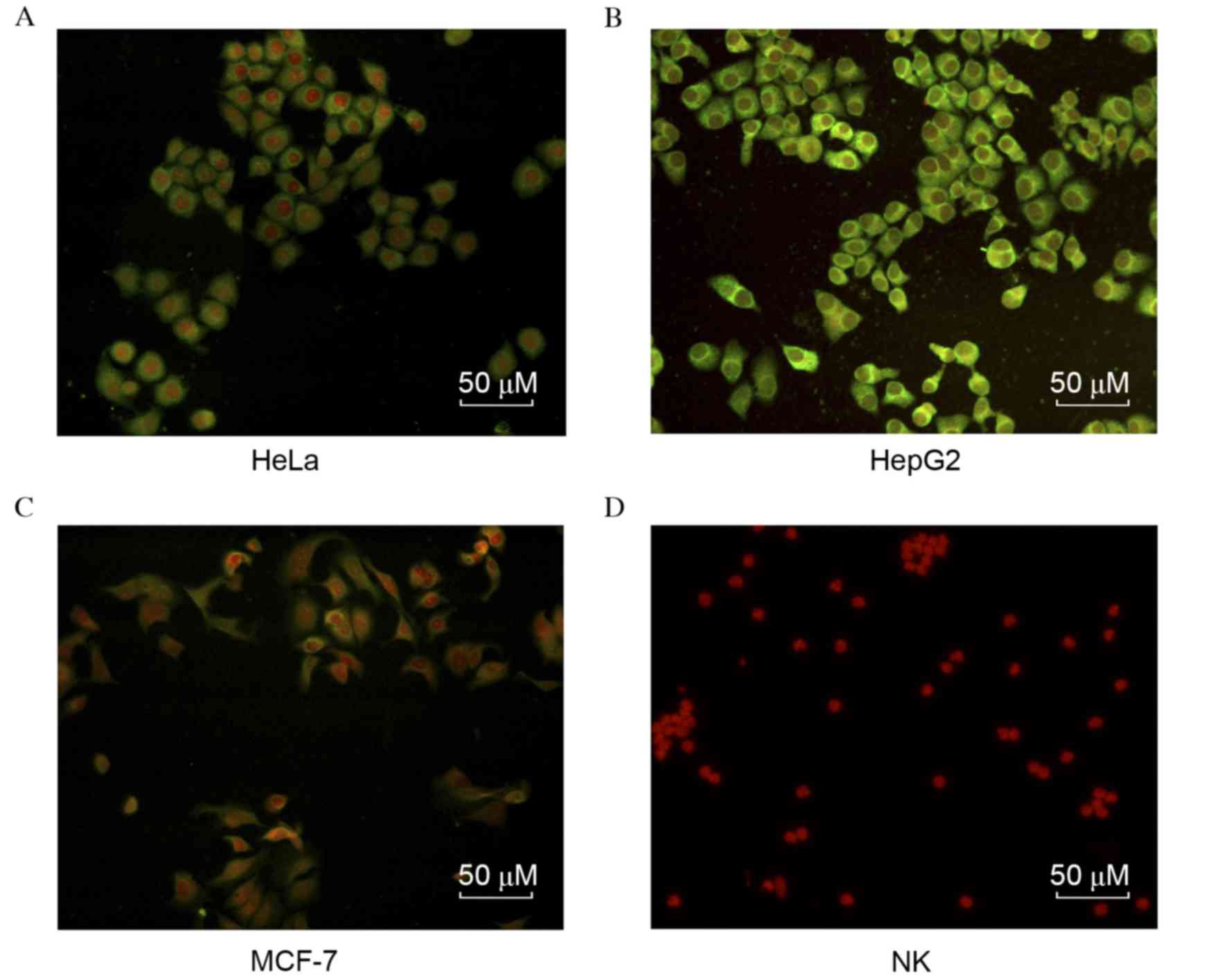
carcinoma cell lines
To identify the specificity of the mAbs against
HAAH-C, the HAAH protein expression levels in each of the three
carcinoma cell lines (HeLa, MCF-7 and HepG2) were evaluated by
immunofluorescence, using the aforementioned C9 mAb. As indicated
in Fig. 3, the anti-HAAH-C mAb
exhibited high affinity in the three carcinoma cell lines. HAAH
expression on the cytomembrane of HepG2 cells was higher than in
the other cell lines; similarly, HAAH expression in HeLa cells was
greater than in MCF-7. As the negative control, human primary NK
cells were observed to have no reaction to the mAb.
NK cell-mediated ADCC
Primary human NK cells were harvested following 21
days of ex vivo expansion. The percentage of NK cells
(CD56+CD3−) within the PBMCs was determined
by flow cytometry, using staining with the CD56-PE and CD3-FITC
mAbs. In one donor sample, the maximal percentage of NK cells
following expansion was 93.18% (Fig.
4A); the mean percentage of NK cells in 10 donor samples was
90.12±4.23% (n=10). With a high specificity, anti-HAAH-C mAb was
able to promote NK cells to combine with the target cells and so
increase the ADCC (Fig. 4B). The
results of the current study demonstrated that, in the presence of
10 µg/ml anti-HAAH-C mAbs, the cytotoxicity of NK cells with regard
to HepG2 was markedly increased, enhanced by 16.25% (50.66±3.07%
increased to 66.91±2.46%; P=0.018). The cytotoxicity rates in HeLa
cells and MCF-7 cells were increased by 13.28% (36.52±3.02 to
49.80±3.95%; P=0.024) and 6.26% (55.45±2.5 to 61.7±2.23%; P=0.013),
respectively. Values are presented as the mean ± SD of four
independent experiments.
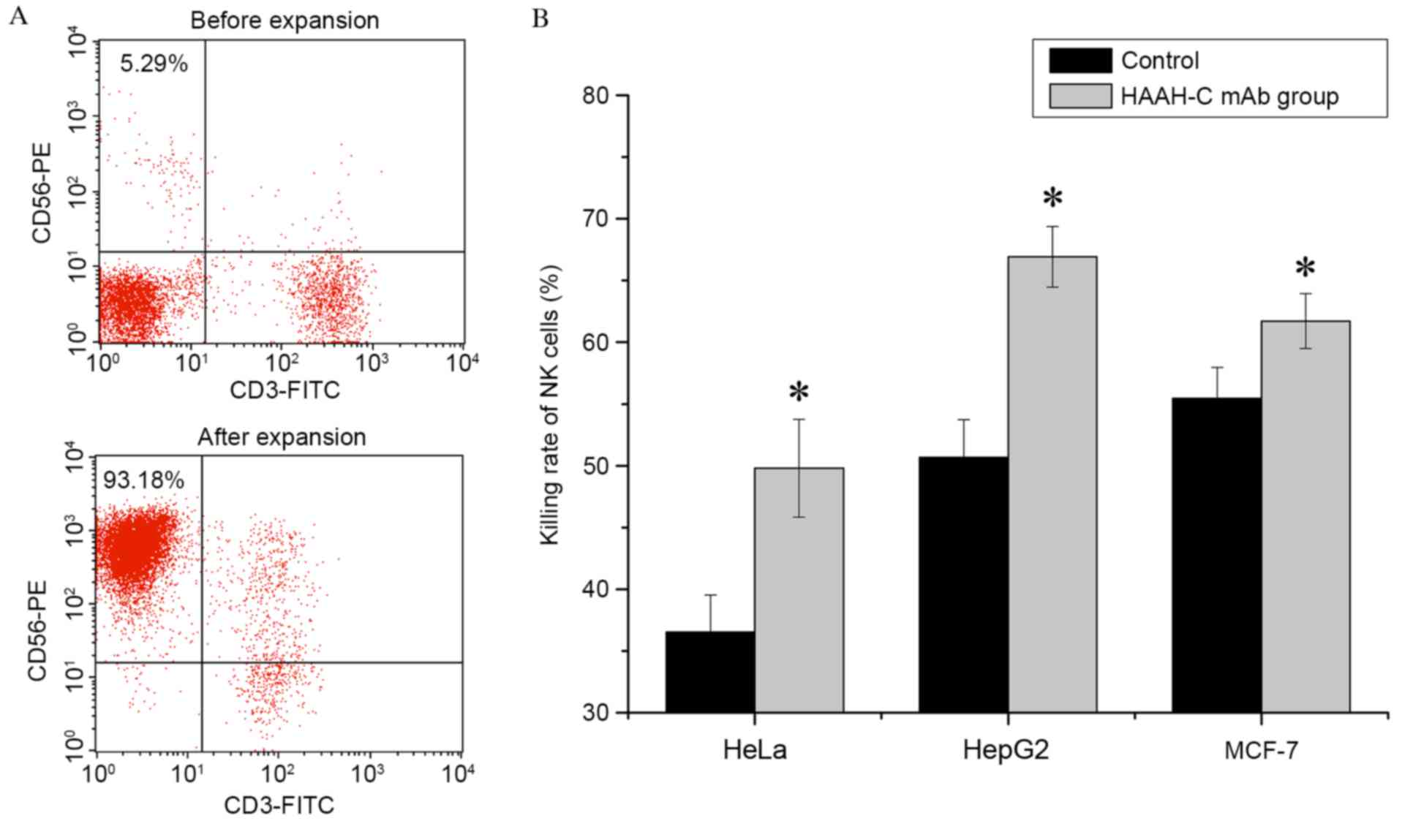 | Figure 4.NK cell expansion in vitro and
ADCC induced by HAAH-C mAb treatment in vitro. (A) PBMCs
were co-cultured with stimulating cells and harvested following a
21-day ex vivo expansion. All pellets were stained with
CD56-PE and CD3-FITC mAbs and analyzed by flow cytometry. The
percentage of NK cells (CD56+CD3−) in the
PBMC population was determined. PBMCs were analyzed by flow
cytometry before and after expansion. The percentage of NK cells
(CD56+CD3−) in the PBMCs was ~5.29% before
expansion and ~93.18% after expansion. (B) In the ADCC assay, HeLa,
HepG2 and MCF-7 tumor cells were mixed with NK cells at ratio of
10:1 in the presence or absence of the HAAH-C mAb (1 µg/ml).
Following a 4-h incubation at 37°C, cell samples were stained with
Cell Counting Kit-8 and the killing rate of the NK cells was
analyzed by a multiscan spectrum. Values are presented as the means
(± standard deviation) of four independent experiments, *P<0.05
vs. the control. HAAH, human aspartyl-(asparaginyl)-β-hydroxylase;
rHAAH-C, recombinant HAAH C-terminal; P. pastoris, Pichia
pastoris; mAbs, monoclonal antibodies; NK, natural killer;
FITC, fluorescein isothiocyanate; PE, phycoerythrin; ADCC,
antibody-dependent cellular cytotoxicity; PBMCs, peripheral blood
mononuclear cells. |
HAAH detection in carcinoma cell
culture supernatant
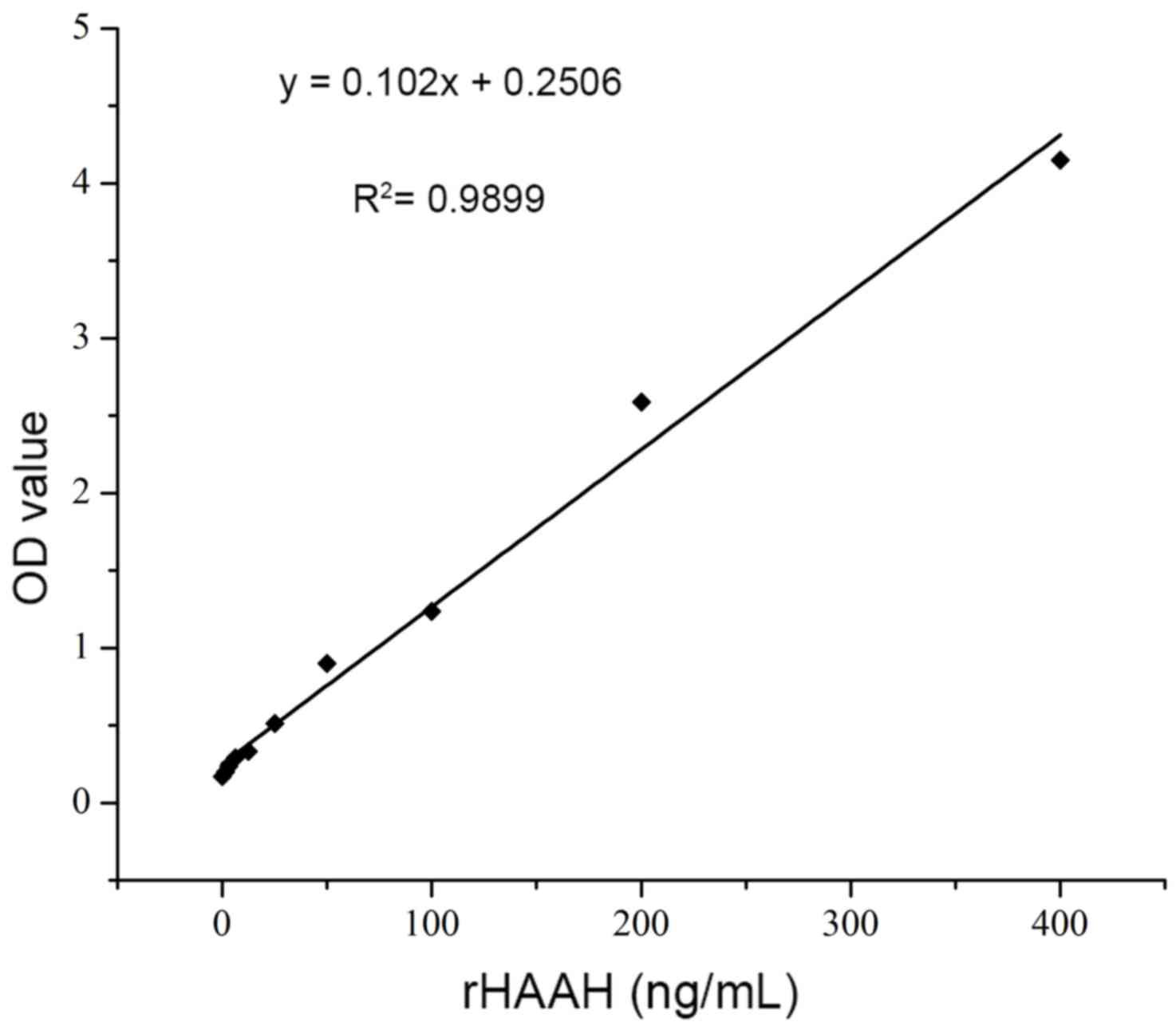
By using a double antibody sandwich ELISA, the
levels of soluble HAAH in the culture supernatant of the carcinoma
cell lines were determined. According to the chessboard reagent
titrations, the optimized concentration of the capture antibody was
10 µg/ml in 100 µl per well. The dilution of the detection antibody
was 1:1,000 (1 ng/ml) and that of the secondary antibody was 1:250.
Recombinant HAAH was prepared in serial dilutions (1.56–400 ng/ml)
to generate a standard curve. The association between the
A450 and the HAAH antigen concentration was obtained as
demonstrated in the following formula: y=0.102x+0.2506,
R2=0.9899; this association exhibited a good linear
response in the concentration range of 10–200 ng/ml (Fig. 5). The cut off value was 2 ng/ml. From
this standard curve, the concentrations of soluble HAAH in the
carcinoma culture supernatant were determined to be 85±35 ng/ml
(HeLa), 202±47 ng/ml (HepG2) and 47±19 ng/ml (MCF-7). The HAAH
protein was not detected in the NK cell supernatant (negative
control).
Discussion
HAAH, a membrane protein with hydroxylation
activity, has been observed to be overexpressed in numerous
malignant neoplasms and has recently been the subject of several
studies (4–7,14,16). A previous study demonstrated that the
HAAH gene was frequently overexpressed in a variety of carcinomas
in Asian patients, by contrast to its relatively low expression in
normal tissues (8). HAAH can also be
detected in the sera of patients with breast, ovarian, lung and
prostate cancers (3–6). As such, the development of an efficient
detection method is critical.
Several hosts, including E. coli and various
mammalian cell lines, have been used to express the recombinant
subtype proteins of HAAH (2,17). A previous investigation also used the
pProEX expression system to express humbug (a subtype of HAAH) in
E. coli (11). One of the
limitations of the expression system in E. coli, was that
the formation of inclusion bodies and its refolding purification
may reduce the recovery yields (18).
To obtain the HAAH-C protein with an improved three-dimensional
structure and ability to prepare a mAb, the P. pastoris
expression system was selected for the current study (19). The procedure for the protein
expression and purification involved three steps. Firstly, a
eukaryotic P. pastoris expression system that was compatible
with the insertion of, and selection for, multiple gene copies was
selected for the HAAH expression (data not shown). Secondly, high
cell-density fermentation in a 10-L bioreactor facilitated the
effective production of the recombinant HAAH protein under
controlled induction conditions, including the pH, temperature and
dissolved O2 concentration. Finally, a two-step
purification procedure was utilized to maintain the functionality
and yield of the rHAAH-C.
In a previous study, the recombinant humbug had been
expressed in E. coli and a mAb against humbug (containing
the HAAH-N terminal) was obtained by co-immunizing mice (8). In the present study, the rHAAH C protein
was expressed using the P. pastoris expression system and
the mAb against this recombinant protein was obtained. Compared
with a previous mAb (11), this novel
anti-HAAH-C mAb had improved activity (a titer of
1.5×104 compared with 1×104 for the
anti-HAAH-N mAb) and was able to combine with the HAAH expressed on
tumor cell membranes. Immunofluorescent cell staining results
indicated that, compared with the low or absent expression in the
primary NK cells, HAAH was overexpressed in the HeLa, MCF-7 and
HepG2 carcinoma cell lines and its distribution pattern was
primarily cytoplasmic, with perinuclear and plasmalemmal
accentuation.
As an activating low-affinity receptor, CD16
(FcgRIIIa) is able to recognize the fragment crystallizable region
of IgG-isotype antibodies. The majority of NK cells express CD16,
through which they recognize and target IgG-coated cells, in a
process termed ADCC (20). It has
been previously reported that IgG1 antibodies are more
ADCC-efficient than IgG2 antibodies (21,22). In
the present study, the prepared HAAH-C mAb was an IgG1-isotype; the
ADCC of NK cells was assayed. The results of the present study
revealed that the HAAH-C mAb increased the ADCC of the NK cells on
HeLa, MCF-7 and HepG2 cells. Among them, the cytotoxicity of NK
cells on HepG2 exhibited a significant increase. This may have been
associated with HepG2 expressing the most HAAH on its cell
membrane.
Carcinoma immunotherapy and chimeric antigen
receptor NK cell immunotherapy has been the subject of numerous
studies in recent years (12,21,23). The
cytotoxicity of NK cells on tumor cells has been utilized to treat
various types of cancer, including breast, ovarian, lung and
prostate, without the adverse effects of radiotherapy or
chemotherapy. The characteristics of the novel anti-HAAH-C mAb,
with increased NK cell ADCC activity, indicate its potential
applications in immunotherapy for liver carcinomas, in addition to
other types of tumors. Furthermore, coupled with the exclusivity of
the surface expression of HAAH on tumor cells, the high affinity of
the anti-HAAH-C mAb for HAAH indicates that it may be developed as
a vehicle for the specific delivery of cytotoxic agents to certain
types of tumor cells and tissues.
In the present study, a double antibody
sandwich-ELISA has been demonstrated to be a promising diagnostic
tool for the detection of the circulating HAAH antigen in the sera
of cancer patients. Thus far, the available commercial ELISA kits
for the detection of HAAH were developed based on a polyclonal
antibody strategy. In the present study, by employing the novel
prepared anti-HAAH-C mAb and the existing anti-HAAH-N mAb, the
double antibody sandwich-ELISA method was developed to detect the
soluble HAAH protein in the culture supernatant of certain
carcinoma cell lines. HepG2 cells were observed to secrete the
majority of the HAAH in the supernatant, 200 ng/ml, whereas HAAH
was barely detectable or undetectable in the supernatant of the NK
cells. The cut off value of the preliminary method developed in the
present study was 2 ng/ml.
The detection of HAAH levels in the sera of tumor
patients remains challenging. A study conducted by Panacea Global
Inc. (24–27) indicated that the mean level of HAAH in
the sera of patients with lung cancer was 18–22 ng/ml. In the sera
of breast, colorectal and prostate cancer patients, this mean level
was 15–19, 24–34 and 17.6–34.6 ng/ml, respectively. The ELISA
method developed in the present study exhibited a linear
association in the concentration range of 10–200 ng/ml, which may
be sufficient to facilitate HAAH detection in patient sera. Further
studies may examine the HAAH levels in cancer patient sera with a
larger number of tissue samples, in order to verify the hypothesis
of the present study and improve the discussed detection
method.
To the best of our knowledge, this is the first
report detailing the preparation of an anti-HAAH-C mAb; the results
of the present study indicate that it may be an effective tool to
use in further studies of HAAH detection, distribution and
function, and that it may be a potential antitumor drug for the
immunotherapy of various types of cancer.
Acknowledgements
The authors acknowledge the financial support from
the National Natural Science Foundation of China (grant nos.
31500688, 11472224 and 81502465), the Fundamental Research Funds
for the Central Universities [grant nos. 3102015ZY099, 3102015BJ
(II)GH09 and 3102016OQD042], and the Basic Research Foundation of
Northwestern Polytechnical University (grant no. JC20110286).
References
|
1
|
Dinchuk JE, Focht RJ, Kelley JA, Henderson
NL, Zolotarjova NI, Wynn R, Neff NT, Link J, Huber RM, Burn TC, et
al: Absence of post-translational aspartyl beta-hydroxylation of
epidermal growth factor domains in mice leads to developmental
defects and an increased incidence of intestinal neoplasia. J Biol
Chem. 277:12970–12977. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Treves S, Feriotto G, Moccagatta L,
Gambari R and Zorzato F: Molecular cloning, expression, functional
characterization, chromosomal localization and gene structure of
junctate, a novel integral calcium binding protein of
sarco(endo)plasmic reticulum membrane. J Biol Chem.
275:39555–39568. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ince N, de la Monte SM and Wands JR:
Overexpression of human aspartyl (asparaginyl) beta-hydroxylase is
associated with malignant transformation. Cancer Res. 60:1261–1266.
2000.PubMed/NCBI
|
|
4
|
Maeda T, Taguchi K, Aishima S, Shimada M,
Hintz D, Larusso N, Gores G, Tsuneyoshi M, Sugimachi K, Wands JR
and de la Monte SM: Clinicopathological correlates of aspartyl
(asparaginyl) beta-hydroxylase over-expression in
cholangiocarcinoma. Cancer Detect Prev. 28:313–318. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lavaissiere L, Jia S, Nishiyama M, de la
Monte S, Stern AM, Wands JR and Friedman PA: Overexpression of
human aspartyl(asparaginyl)beta-hydroxylase in hepatocellular
carcinoma and cholangiocarcinoma. J Clin Invest. 98:1313–1323.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sepe PS, Lahousse SA, Gemelli B, Chang H,
Maeda T, Wands JR and de la Monte SM: Role of the
aspartyl-asparaginyl-beta-hydroxylase gene in neuroblastoma cell
motility. Lab Invest. 82:881–891. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dinchuk JE, Henderson NL, Burn TC, Huber
R, Ho SP, Link J, O'Neil KT, Focht RJ, Scully MS, Hollis JM, et al:
Aspartyl beta-hydroxylase (Asph) and an evolutionarily conserved
isoform of Asph missing the catalytic domain share exons with
junctin. J Biol Chem. 275:39543–39554. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang H, Song K, Xue T, Xue XP, Huyan T,
Wang W and Wang H: The distribution and expression profiles of
human Aspartyl/Asparaginyl beta-hydroxylase in tumor cell lines and
human tissues. Oncol Rep. 24:1257–1264. 2010.PubMed/NCBI
|
|
9
|
Xue T, Su J, Li H and Xue X: Evaluation of
HAAH/humbug quantitative detection in the diagnosis of
hepatocellular carcinoma. Oncol Rep. 33:329–337. 2015.PubMed/NCBI
|
|
10
|
Huyan T, Li Q, Ye LJ, Yang H, Xue XP,
Zhang MJ, Huang QS, Yin DC and Shang P: Inhibition of human natural
killer cell functional activity by human aspartyl β-hydroxylase.
Int Immunopharmacol. 23:452–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xue T, Xue XP, Huang QS, Wei L, Sun K and
Xue T: Monoclonal antibodies against human aspartyl (asparaginyl)
beta-hydroxylase developed by DNA immunization. Hybridoma
(Larchmt). 28:251–257. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sharp PM and Li WH: The codon adaptation
index-a measure of directional synonymous codon usage bias, and its
potential applications. Nucleic Acids Res. 15:1281–1295. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Q, Mei Q, Huyan T, Xie L, Che S, Yang
H, Zhang M and Huang Q: Effects of simulated microgravity on
primary human NK cells. Astrobiology. 13:703–714. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xian ZH, Zhang SH, Cong WM, Yan HX, Wang K
and Wu MC: Expression of aspartyl beta-hydroxylase and its
clinicopathological significance in hepatocellular carcinoma. Mod
Pathol. 19:280–286. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo Y, Terkawi MA, Jia H, Aboge GO, Goo
YK, Cao S, Li Y, Yu L, Ooka H, Kamyingkird K, et al: A double
antibody sandwich enzyme-linked immunosorbent assay for detection
of secreted antigen 1 of Babesia microti using hamster model. Exp
Parasitol. 130:178–182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maeda T, Sepe P, Lahousse S, Tamaki S,
Enjoji M, Wands JR and de la Monte SM: Antisense
oligodeoxynucleotides directed against aspartyl (asparaginyl)
beta-hydroxylase suppress migration of cholangiocarcinoma cells. J
Hepatol. 38:615–622. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee JH: Overexpression of humbug promotes
malignant progression in human gastric cancer cells. Oncol Rep.
19:795–800. 2008.PubMed/NCBI
|
|
18
|
Baneyx F: Recombinant protein expression
in Escherichia coli. Curr Opin Biotechnol. 10:411–421. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brake AJ, Merryweather JP, Coit DG,
Heberlein UA, Masiarz FR, Mullenbach GT, Urdea MS, Valenzuela P and
Barr PJ: Alpha-factor-directed synthesis and secretion of mature
foreign proteins in Saccharomyces cerevisiae. Proc Natl Acad Sci
USA. 81:4642–4646. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oppenheim DE, Spreafico R, Etuk A, Malone
D, Amofah E, Peña-Murillo C, Murray T, McLaughlin L, Choi BS, Allan
S, et al: Glyco-engineered anti-EGFR mAb elicits ADCC by NK cells
from colorectal cancer patients irrespective of chemotherapy. Br J
Cancer. 110:1221–1227. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nimmerjahn F and Ravetch JV: Divergent
immunoglobulin g subclass activity through selective Fc receptor
binding. Science. 310:1510–1512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schneider-Merck T, van Bueren JJ Lammerts,
Berger S, Rossen K, van Berkel PH, Derer S, Beyer T, Lohse S,
Bleeker WK, Peipp M, et al: Human IgG2 antibodies against epidermal
growth factor receptor effectively trigger antibody-dependent
cellular cytotoxicity but, by contrast to IgG1, only by cells of
myeloid lineage. J Immunol. 184:512–520. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen Y, Wang Y, Zhuang Y, Zhou F and Huang
L: Mifepristone increases the cytotoxicity of uterine natural
killer cells by acting as a glucocorticoid antagonist via ERK
activation. PLoS One. 7:e364132012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Panacea Global, . A New and Sensitive
Diagnostic Test for the Detection of Breast Cancer-BC Detect.
http://www.panaceaglobalinc.com/edit/files/pdfs/bc_detect/35-bcd-pub.pdf
|
|
25
|
Panacea Global, . A New and Sensitive
Diagnostic Test for the Detection of Colorectal Cancer-CC Detect.
http://www.panaceaglobalinc.com/edit/files/pdfs/cc_detect/36ccd-pub.pdf
|
|
26
|
Panacea Global, . A New and Sensitive
Diagnostic Test for the Detection of Lung Cancer-LC Detect.
http://www.panaceaglobalinc.com/edit/files/pdfs/lc_detect/37-lcd-pub.pdf
|
|
27
|
Panacea Global, . A New and Sensitive
Diagnostic Test for the Detection of Prostate Cancer-PC Detect.
http://www.panaceaglobalinc.com/edit/files/pdfs/pc_detect/38-pcd-pub.pdf
|